Was ist es ausgelassen systemisch Lupus erythematodes?
Bullöser systemischer Lupus erythematodes (SLE) ist a Autoimmun subepidermal bullöse Erkrankung, die bei Patienten mit SLE auftritt. Kann mit verbunden sein Antikörper gegen Typ VII Kollagen.
Bullous SLE wird auch als bullous bezeichnet. Eruption von LES und vesikulobullös SIE.
Bullöser systemischer Lupus erythematodes
LES ausgelassen
Wer bekommt bullösen systemischen Lupus erythematodes?
das Vorfall Der Anteil bullöser SLE wurde in Frankreich und Singapur auf 0,22 und 0,26 Fälle pro Million pro Jahr geschätzt. In einer großen Kohorte von Seren von Patienten mit genommen immunobullös Störungen, 1-2%, wurden als bullöser SLE identifiziert [1,2].
Wie SLE wurde auch bei Frauen afrikanischer Herkunft in den Dreißigern am häufigsten über bullösen SLE berichtet. Bullöser SLE tritt jedoch in allen Altersgruppen, Geschlechtern und Ethnien auf.
Das bullöse LES kann die Präsentation sein Zeichen von SLE bei etwa einem Drittel der Patienten. In den meisten Fällen wurde SLE jedoch bereits vor einigen Jahren diagnostiziert.
Was verursacht bullösen systemischen Lupus erythematodes?
Bullous SLE wird gemäß in drei Typen eingeteilt Immunhistochemie.
- Bullöser SLE vom Typ I ist der häufigste Typ und wird durch Autoantikörper definiert, die gegen Kollagen vom Typ VII gerichtet sind, insbesondere gegen Nichtkollagen.Kollagen Domänen vom Typ 1 und Typ 2 (NC1 und NC2). Die NC1-Domäne spielt eine wichtige Rolle bei der Aufrechterhaltung der Struktur der dermal-epidermal Kreuzung (DEJ) [3–5].
- Bei bullösem SLE vom Typ II sind Antikörper gegen andere gerichtet Antigene DEJ, einschließlich BP130, BP280 und Laminin 5 oder 6.
- Typ III bullöser SLE ist durch Färbung in der Epidermis mit oder ohne Färbung in der Dermis.
Die drei Typen können klinisch nicht unterschieden werden.
Prädisponierende Faktoren sind Arzneimittelnebenwirkungen zu Hydralazin, Penicillamin, Methimazol; und UV-Exposition (UV) Strahlung B.
Typ I bullöser SLE ähnelt der Epidermolysis bullosa bullosa (EBA) darin, dass beide haben Autoantikörper Kollagen Typ VII in indirekter Immunfluoreszenz [6]. Bullöse SLE und EBA unterscheiden sich jedoch klinisch, und es wurde nun gezeigt, dass IgG-Autoantikörper unterschiedlich sind.
Was sind die klinischen Merkmale des bullösen systemischen Lupus erythematodes?
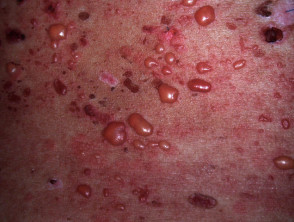
In LES ausgelassen, angespannt Vesikel, Bullae und Erosionen entstehen normal oder erythematös Haut, in der Regel an Orten, die der Sonne ausgesetzt sind.
- Das Gesicht ist oft betroffen; Schleimhaut Oberflächen können ebenfalls beteiligt sein.
- Der Beginn des bullösen Ausbruchs korreliert nicht mit der Aktivität der systemischen Erkrankung.
- Die Blasen können klar sein oder hämorrhagisch Inhalt.
- Die Blasen heilen ohne Narben.
- Pruritus es fehlt oder ist mild.
- Milia Sie sind kein herausragendes Merkmal.
- Erythematös stornieren Urtikaria Platten kann entstehen.
Was sind die Komplikationen des bullösen systemischen Lupus erythematodes?
Die Komplikationen des bullösen SLE sind die des zugrunde liegenden SLE. das Entwicklung von bullösem SLE wird oft mit einer Lupusfackel in Verbindung gebracht Nephritis.
Wie wird bullöser systemischer Lupus erythematodes diagnostiziert?
Untersuchungen sind notwendig, um bullösen SLE von SLE mit a zu unterscheiden begleitend Blasenstörung.
Histopathologisch Die Ergebnisse sehen gleich aus Dermatitis Herpetiformis, da beide zeigen:
- Subepidermale Teilung
- EIN Neutrophile vorherrschend infiltrieren in der oberen Dermis
- MikrokapillareAbszesse
- Leukozytoklasis
- Mucin Erklärung in der Dermis.
Basal Deckel Vakuolisierung, charakteristisch für andere Formen von Haut- LE ist nicht vorhanden.
Die direkte Funktion Immunfluoreszenz (DIF) Befund in a perilesional Haut Biopsie von LES bulloso ist linear oder körnig Immunoglobulin (Ig) G, IgM, IgA oder C3 am dermoepidermalen Übergang (DEJ). Bullöser SLE vom Typ I ist typischerweise mit dem linearen Muster verbunden.
Serum Autoantikörper nachgewiesen durch indirekten Immunfluoreszenztest oder EnzymDer verknüpfte Immunosorbens-Assay (ELISA) ist gegen Typ VII-Kollagen in bullösem Typ I-SLE. BP 230, BP 180 oder Laminin 5 oder 6 werden in bullösem Typ II-SLE nachgewiesen.
DIF-Zahnmusteranalyse Mikroskopie unterscheidet gewebegebundene Autoantikörper gegen Kollagen Typ VII, bei dem ein U-gezacktes Muster beobachtet wird, von allen anderen Anti-DEJ-Antikörpern, bei denen ein N-gezacktes Muster beobachtet wird [7].
Diagnosekriterien für bullösen SLE erfordern:
- Eine Diagnose von SLE basierend auf dem American College of Rheumatologie Kriterien
- Ein erworbener vesikulärer bullöser Ausschlag
- Histopathologie Hinweise auf eine subepidermale Blase und ein überwiegend neutrophiles dermales Infiltrat, das der Dermatitis herpetiformis ähnelt
- DIF-Mikroskopie zeigt lineare oder körnige Ablagerungen von Immunglobulin (Ig) G oder IgM und häufig IgA in der Basalmembran Zone
- Indirekte positive oder negative Immunfluoreszenz für zirkulierende Autoantikörper im Bereich der Basalmembran.
Welches ist das Differenzialdiagnose für bullösen systemischen Lupus erythematodes?
Die Differentialdiagnose für bullösen SLE lautet bullöser SLE. Andere Blasenzustände, die bei SLE auftreten können, sind:
- Bullöser Drogenausbruch: Bandförmige Ablagerungen von IgG, IgM und IgA können vorhanden sein; Es werden jedoch keine linearen oder körnigen Ablagerungen beobachtet
- Die pemphigoidale Gruppe von Krankheiten: bullöses Pemphigoid, lineare IgA-Krankheit, Epidermolysis bullosa und Dermatitis herpetiformis.
- Haut Verletzungen von sehr aktivem SLE: Diese können aufgrund einer starken Vakuolisierung im JED selten Blasen bilden.
Was ist die Behandlung für bullösen systemischen Lupus erythematodes?
Dapson in Dosen von 1,0 bis 1,5 mg / kg / Tag wird als Behandlung der Wahl angesehen. Es ist bei der überwiegenden Mehrheit der Patienten wirksam und führt innerhalb weniger Tage bis zu einigen Wochen zu einer raschen klinischen Besserung. Diese schnelle Reaktion unterscheidet bullösen SLE von SLE mit einer begleitenden autoimmunen bullösen Erkrankung.
Wenn Dapson versagt, werden Prednisolon (systemische Steroide) und Azathioprin verwendet. Fälle von Patienten, die erfolgreich mit Methotrexat oder Rituximab behandelt wurden, wurden berichtet [8–11].
Was ist das Ergebnis eines bullösen systemischen Lupus erythematodes?
Das bullöse LES ist vorübergehend in den meisten Fällen und in der Regel vollständig ohne weitere Schübe, unabhängig von der systemischen Krankheitsaktivität. Es löst sich normalerweise ohne Milie oder Narbenbildung auf, kann aber Hypo- oder Hyper- hinterlassenPigmentierung.