Lupus Eritematoso Sistémico Bulloso: Definición y Características Fundamentales
El lupus eritematoso sistémico bulloso (LES bulloso) es una enfermedad ampollosa autoinmune subepidérmica que se desarrolla en pacientes que ya presentan un diagnóstico establecido de Lupus Eritematoso Sistémico (LES). Esta manifestación específica a menudo se vincula con la actividad de anticuerpos que tienen como diana el colágeno tipo VII, una proteína esencial para la piel.
Esta variante clínica también es reconocida bajo denominaciones alternativas, tales como erupción bullosa del LES o LES vesiculobulloso, haciendo referencia a su naturaleza formadora de ampollas.
Entendiendo el Lupus Eritematoso Sistémico Ampolloso
LES bulloso
Perfil Demográfico de los Afectados por LES Bulloso
La incidencia del LES bulloso se considera baja; estimaciones recopiladas en regiones como Francia y Singapur sugieren cifras anuales entre 0,22 y 0,26 casos por cada millón de habitantes. Adicionalmente, un examen detallado de sueros obtenidos de pacientes con diversas afecciones inmunobullosas reveló que solo entre el 1% y el 2% de estos casos correspondían específicamente al LES ampolloso [1,2].
Al igual que ocurre con el LES en su forma general, esta manifestación cutánea bullosa se observa con mayor preponderancia en mujeres de ascendencia africana que se encuentran en la tercera década de vida. Sin embargo, es crucial recordar que el LES bulloso puede manifestarse independientemente de la edad, el sexo o la etnia del individuo.
Para aproximadamente un tercio de las personas diagnosticadas, el LES bulloso emerge como el primer signo de lupus sistémico. Aun así, en la mayoría de los pacientes, el diagnóstico de LES ya se había establecido previamente, pudiendo haber transcurrido incluso varios años antes de la aparición de las lesiones ampollares características.
Clasificación Inmunocitoquímica y Etiología del LES Bulloso
El LES bulloso se estratifica en tres subtipos predominantes, determinados por el patrón específico de hallazgos inmunohistoquímicos:
- El LES bulloso tipo I es la presentación más común. Se identifica por la respuesta autoinmune dirigida contra el colágeno tipo VII, centrándose particularmente en los dominios no colágenos tipo 1 y tipo 2 (NC1 y NC2). El dominio NC1 es fundamental, ya que sostiene la estructura de la unión dérmico-epidérmica (DEJ) [3–5].
- En el LES bulloso tipo II, los anticuerpos atacan otros antígenos স্থানীয়izados también en la DEJ. Estos incluyen BP130, BP280, o las proteínas lamininas 5 o 6.
- El LES bulloso tipo III se caracteriza por presentar una tinción positiva en la unión dermoepidérmica.
Comprender estas clasificaciones ayuda a los especialistas a determinar el mecanismo patogénico subyacente más probable en cada caso de lupus eritematoso sistémico bulloso.
- epidermis, con o sin tinción adicional en la dermis.
Clínicamente, resulta imposible distinguir entre estos tres subtipos de LES ampolloso.
Factores que pueden impulsar su desarrollo incluyen reacciones adversas a fármacos, como las inducidas por hidralazina, penicilamina o metimazol, así como la exposición a la radiación ultravioleta B (UV).
Es importante señalar que el LES ampolloso tipo I comparte rasgos con la penfigoide ampollosa (EBA), ya que ambas patologías se vinculan a la presencia de autoanticuerpos.
Para abordar de manera efectiva y comprender las implicaciones de esta compleja afección autoinmune, es fundamental la consulta con especialistas en dermatología que estén al tanto de las investigaciones más recientes sobre enfermedades ampollosas.
Diagnóstico e Implicaciones Clínicas del Lupus Eritematoso Sistémico Ampolloso
El diagnóstico diferencial del lupus eritematoso sistémico ampolloso (LES ampolloso) requiere frecuentemente diferenciarlo de otros trastornos ampollosos autoinmunes, como la penfigoide ampollosa (EBA). Las pruebas mediante inmunofluorescencia indirecta ```html Técnica de Inmunofluorescencia Indirecta (IFI): Fundamentos y Aplicaciones Diagnósticas ¿Qué es la Inmunofluorescencia Indirecta (IFI)? La inmunofluorescencia indirecta, también denominada inmunofluorescencia secundaria, es una técnica esencial en el laboratorio clínico empleada para la detección de autoanticuerpos presentes en el suero del paciente. Su principal utilidad reside en el diagnóstico de enfermedades autoinmunes de tipo ampolloso. • Anticuerpos primarios (no etiquetados), que pueden actuar como marcadores para condiciones como el más son esenciales para detectar anticuerpos dirigidos contra el colágeno tipo VII [6]. Si bien el LES ampolloso y la EBA pueden mostrar similitudes histológicas, se ha establecido que los autoanticuerpos IgG involucrados son molecularmente distintos en cada patología.
```html Técnica de Inmunofluorescencia Indirecta (IFI): Fundamentos y Aplicaciones Diagnósticas ¿Qué es la Inmunofluorescencia Indirecta (IFI)? La inmunofluorescencia indirecta, también denominada inmunofluorescencia secundaria, es una técnica esencial en el laboratorio clínico empleada para la detección de autoanticuerpos presentes en el suero del paciente. Su principal utilidad reside en el diagnóstico de enfermedades autoinmunes de tipo ampolloso. • Anticuerpos primarios (no etiquetados), que pueden actuar como marcadores para condiciones como el más son esenciales para detectar anticuerpos dirigidos contra el colágeno tipo VII [6]. Si bien el LES ampolloso y la EBA pueden mostrar similitudes histológicas, se ha establecido que los autoanticuerpos IgG involucrados son molecularmente distintos en cada patología.
Presentación Clínica del Lupus Eritematoso Sistémico Ampolloso
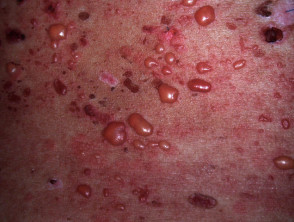
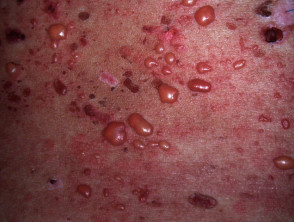
En el LES ampolloso, la manifestación cutánea se caracteriza por la aparición de vesículas tensas, bullas y erosiones localizadas sobre áreas de piel que pueden lucir normales o presentar un fondo eritematoso, afectando predominantemente las zonas fotoexpuestas.
- La cara es una localización frecuente; las superficies de la mucosa también pueden estar afectadas.
- La aparición del exantema ampolloso no se correlaciona con la actividad sistémica del lupus.
- El contenido vesicular puede ser transparente o hemorrágico.
- Las ampollas evolucionan hacia la curación sin generar cicatrices permanentes.
- El prurito (picazón) generalmente está ausente o es leve.
- La formación de milia no es una característica prominente de la presentación.
- Es posible observar el surgimiento de placas de urticaria anular eritematosa.
Complicaciones Relacionadas con el Lupus Eritematoso Sistémico Ampolloso
Las complicaciones primarias del LES ampolloso suelen derivar directamente de la afectación sistémica subyacente. Es crucial notar que la irrupción del LES ampolloso se asocia frecuentemente con un episodio de nefritis lúpica activa.
Herramientas Diagnósticas para el Lupus Eritematoso Sistémico Ampolloso
Se requieren evaluaciones clínicas detalladas para establecer la diferencia entre el LES ampolloso puro y la coexistencia de LES junto con un trastorno ampolloso concomitante.
Los hallazgos histopatológicos muestran paralelismos con la dermatitis Comprendiendo la Dermatitis: Definición, Causas y Tipos Comunes La dermatitis abarca un conjunto de afecciones inflamatorias que se manifiestan a través de cambios específicos en la epidermis, manifestándose frecuentemente como picazón intensa. Esta condición es notablemente común, afectando a cerca de una quinta parte de la población en algún momento de sus vidas. Debido a su etiología diversa, la dermatitis presenta múltiples patrones de manifestación clínica. Los términos "dermatitis" y más herpetiforme, dado que ambas condiciones comparten los siguientes atributos histológicos:
Comprendiendo la Dermatitis: Definición, Causas y Tipos Comunes La dermatitis abarca un conjunto de afecciones inflamatorias que se manifiestan a través de cambios específicos en la epidermis, manifestándose frecuentemente como picazón intensa. Esta condición es notablemente común, afectando a cerca de una quinta parte de la población en algún momento de sus vidas. Debido a su etiología diversa, la dermatitis presenta múltiples patrones de manifestación clínica. Los términos "dermatitis" y más herpetiforme, dado que ambas condiciones comparten los siguientes atributos histológicos:
- Presencia de separación a nivel subepidérmico.
- Un infiltrado dérmico superior compuesto predominantemente por neutrófilos.
- Identificación de microabscesos micropapilares.
- Signos de leucocitoclasia.
La correcta identificación de las características clínicas y los marcadores inmunológicos es vital para diferenciar el LES ampolloso de otras dermatosis con tendencia a la formación de ampollas, asegurando así un tratamiento dirigido y efectivo.
- Depósito de mucina en la dermis.
La vacuolización de la capa basal, una característica distintiva en otras formas de lupus eritematoso cutáneo, no se identifica en este contexto específico.
El hallazgo primordial mediante inmunofluorescencia directa ```html Comprensión de la Técnica de Inmunofluorescencia Directa (DIF) La inmunofluorescencia directa (DIF) es una técnica de laboratorio fundamental empleada para el diagnóstico preciso de patologías cutáneas, renales y de otros sistemas orgánicos. También es conocida como prueba fluorescente inmunitaria directa o como el método de inmunofluorescencia primario. El procedimiento de DIF implica la aplicación de anticuerpo-fluoróforo conjugado moléculas directamente sobre muestras de tejido obtenidas mediante biopsias del paciente. Estos más (DIF) en una biopsia de piel
```html Comprensión de la Técnica de Inmunofluorescencia Directa (DIF) La inmunofluorescencia directa (DIF) es una técnica de laboratorio fundamental empleada para el diagnóstico preciso de patologías cutáneas, renales y de otros sistemas orgánicos. También es conocida como prueba fluorescente inmunitaria directa o como el método de inmunofluorescencia primario. El procedimiento de DIF implica la aplicación de anticuerpo-fluoróforo conjugado moléculas directamente sobre muestras de tejido obtenidas mediante biopsias del paciente. Estos más (DIF) en una biopsia de piel ```html Guía Completa sobre la Biopsia de Piel: Definición y Tipos ¿Qué es una Biopsia de Piel y Cómo se Realiza? Una biopsia de piel implica la extracción minuciosa de una muestra de tejido cutáneo para su posterior análisis. Generalmente, este procedimiento se realiza bajo anestesia local. Se administra una inyección del anestésico directamente en el área a tratar, lo cual puede causar una sensación punzante transitoria. Finalizado el muestreo, más perilesional, cuando se trata del LES ampolloso, consiste en el depósito lineal o granular de inmunoglobulina (Ig) G, IgM, IgA o C3 en la unión dermoepidérmica (DEJ). El LES ampolloso tipo I se vincula típicamente con este patrón lineal de deposición.
```html Guía Completa sobre la Biopsia de Piel: Definición y Tipos ¿Qué es una Biopsia de Piel y Cómo se Realiza? Una biopsia de piel implica la extracción minuciosa de una muestra de tejido cutáneo para su posterior análisis. Generalmente, este procedimiento se realiza bajo anestesia local. Se administra una inyección del anestésico directamente en el área a tratar, lo cual puede causar una sensación punzante transitoria. Finalizado el muestreo, más perilesional, cuando se trata del LES ampolloso, consiste en el depósito lineal o granular de inmunoglobulina (Ig) G, IgM, IgA o C3 en la unión dermoepidérmica (DEJ). El LES ampolloso tipo I se vincula típicamente con este patrón lineal de deposición.
Los autoanticuerpos séricos, detectados por inmunofluorescencia indirecta o mediante ensayos inmunoabsorbentes ligados a enzimas (ELISA), muestran reactividad específica. En el LES ampolloso tipo I, estos anticuerpos se dirigen contra el colágeno de tipo VII. Para el LES ampolloso tipo II, se detectan anticuerpos frente a BP 230, BP 180, o laminina 5 o 6.
El análisis del patrón de borde (dentado) de DIF mediante microscopía...
La integración precisa de estos hallazgos clínicos y confirmatorios mediante pruebas es crucial para establecer un diagnóstico acertado de lupus eritematoso ampolloso y distinguirlo de otras condiciones que presentan sintomatología similar.
Es fundamental diferenciar la unión de autoanticuerpos al colágeno tipo VII, que exhibe un patrón dentado con morfología de "U", de otros anticuerpos anti-DEJ (unión dermoepidérmica), donde se observa un patrón dentado en forma de "N" [7].
Los criterios diagnósticos requeridos para confirmar el diagnóstico de Lupus Eritematoso Sistémico (LES) ampolloso son los siguientes:
- Ya debe existir un diagnóstico establecido de LES fundamentado en los criterios definidos por el American College of Reumatología.
- Presencia confirmada de una erupción vesiculobullosa de naturaleza adquirida.
- Evidencia histopatológica que demuestre una ampolla subepidérmica junto con un infiltrado dérmico dominado por neutrófilos, asemejándose a una dermatitis herpetiforme
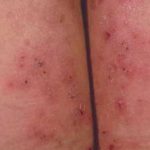 Comprendiendo la Dermatitis Herpetiforme: Causas, Síntomas y Clínica ¿Qué es la Dermatitis Herpetiforme? La dermatitis herpetiforme (DH) es una enfermedad persistente y poco frecuente del grupo inmunobulloso, estrechamente vinculada a la enfermedad celíaca (enteropatía sensible al gluten). Su denominación, "herpetiforme", proviene de la tendencia de las ampollas a manifestarse en agrupaciones que evocan al herpes simplex. Es crucial destacar que, a pesar de esta similitud visual, la dermatitis herpetiforme no más.
Comprendiendo la Dermatitis Herpetiforme: Causas, Síntomas y Clínica ¿Qué es la Dermatitis Herpetiforme? La dermatitis herpetiforme (DH) es una enfermedad persistente y poco frecuente del grupo inmunobulloso, estrechamente vinculada a la enfermedad celíaca (enteropatía sensible al gluten). Su denominación, "herpetiforme", proviene de la tendencia de las ampollas a manifestarse en agrupaciones que evocan al herpes simplex. Es crucial destacar que, a pesar de esta similitud visual, la dermatitis herpetiforme no más. - El estudio de microscopía de inmunofluorescencia directa (DIF) debe revelar depósitos lineales o granulares de inmunoglobulina (Ig) G o IgM, y habitualmente IgA, localizados en la zona de la membrana basal.
- Los resultados obtenidos mediante inmunofluorescencia indirecta deben ser positivos o negativos para detectar autoanticuerpos circulantes dirigidos contra la zona de la membrana basal.
Diagnóstico Diferencial Clave del Lupus Eritematoso Sistémico Ampolloso
El principal parangón diagnóstico del LES ampolloso es, intrínsecamente, el LES que desarrolla formación de ampollas. Sin embargo, existen otras patologías con presentación ampollosa que pueden ocurrir simultáneamente o manifestarse en pacientes con LES establecidas:
- Erupción ampollosa de origen farmacológico: Aunque es posible observar depósitos en banda de IgG, IgM e IgA, en estos casos no se presentan los depósitos lineales o granulares distintivos del LES ampolloso.
- El grupo diverso de enfermedades que componen el penfigoide: Esto abarca específicamente el penfigoide ampolloso
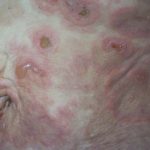 Que es bulloso penfigoide?El penfigoide bulloso es un autoinmune subepidérmico enfermedad ampollosa.¿Quién contrae penfigoide bulloso?El penfigoide ampolloso a menudo se presenta en personas mayores de 80 años y afecta principalmente a personas mayores de 50 años. Puede ocurrir en adultos más jóvenes, pero el penfigoide ampollar en bebés y niños es raro.• El penfigoide ampolloso ocurre por igual en hombres y mujeres. • Existe una asociación con humanos leucocito antígeno más, la enfermedad por IgA lineal, la epidermólisis ampollosa
Que es bulloso penfigoide?El penfigoide bulloso es un autoinmune subepidérmico enfermedad ampollosa.¿Quién contrae penfigoide bulloso?El penfigoide ampolloso a menudo se presenta en personas mayores de 80 años y afecta principalmente a personas mayores de 50 años. Puede ocurrir en adultos más jóvenes, pero el penfigoide ampollar en bebés y niños es raro.• El penfigoide ampolloso ocurre por igual en hombres y mujeres. • Existe una asociación con humanos leucocito antígeno más, la enfermedad por IgA lineal, la epidermólisis ampollosa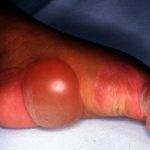 Epidermólisis Ampollosa: Un Trastorno Genético de la Piel La Epidermólisis Ampollosa (EB) es un complejo grupo de afecciones hereditarias caracterizadas por la propensión extrema de la piel y las membranas mucosas a formar ampollas y lesiones (lesiones). Estas vesículas cutáneas aparecen típicamente en zonas sujetas a fricción o mínimo trauma, como las manos y los pies. Sin embargo, en algunos subtipos genéticos, las ampollas pueden manifestarse internamente, afectando órganos como más y la dermatitis herpetiforme.
Epidermólisis Ampollosa: Un Trastorno Genético de la Piel La Epidermólisis Ampollosa (EB) es un complejo grupo de afecciones hereditarias caracterizadas por la propensión extrema de la piel y las membranas mucosas a formar ampollas y lesiones (lesiones). Estas vesículas cutáneas aparecen típicamente en zonas sujetas a fricción o mínimo trauma, como las manos y los pies. Sin embargo, en algunos subtipos genéticos, las ampollas pueden manifestarse internamente, afectando órganos como más y la dermatitis herpetiforme. - Lesiones cutáneas de LES con actividad muy elevada: Es un escenario infrecuente, pero las lesiones pueden ampollarse secundariamente a la extensa vacuolización que tiene lugar en la unión dermoepidérmica (DEJ).
Estrategias de Tratamiento para el Lupus Eritematoso Sistémico Ampolloso
La dapsona```html Información Esencial sobre la Dapsona: Usos, Monitoreo y Efectos Secundarios La dapsona, un antibiótico perteneciente a la clase de las sulfonas, ha sido un tratamiento establecido y disponible durante muchos años, especialmente para la lepra. En el contexto de Nueva Zelanda, este medicamento se presenta comúnmente en tabletas de 25 mg y 100 mg. Usos Dermatológicos de la Dapsona La dapsona es fundamental en el manejo de diversas afecciones más, administrada en rangos de 1.0 a 1.5 mg/kg/día, se establece como el tratamiento de primera línea preferente. Este principio activo demuestra gran eficacia en la gran mayoría de los afectados, logrando una rápida mejoría clínica que se manifiesta en cuestión de días o pocas semanas. Esta respuesta terapéutica acelerada es vital, ya que ayuda a diferenciar el LES ampolloso de una enfermedad ampollosa autoinmune concurrente al LES.
En aquellos escenarios clínicos donde no se observa respuesta adecuada a la dapsona, se emplean como alternativas la prednisolona (esteroides sistémicos) y la azatioprina. Además, existen reportes documentados con éxito de pacientes tratados utilizando metotrexato o rituximabComprendiendo el Rituximab: Mecanismo de Acción y Linfocitos B El rituximab es un medicamento biológico fundamental utilizado primordialmente para el tratamiento del linfoma. Recientemente, ha demostrado ser valioso en el manejo de diversas afecciones cutáneas graves. De hecho, en 2018, la FDA autorizó su uso para el pénfigo vulgar de moderado a grave, una enfermedad inmunobullosa de la piel. Es crucial notar que el rituximab conlleva riesgos de efectos secundarios más [8–11].
Pronóstico Clínico del Lupus Eritematoso Sistémico Ampolloso
En la mayoría de los casos diagnosticados, el LES bulloso exhibe un curso transitorio y tiende a resolverse de manera completa, incluso si la actividad de la enfermedad sistémica subyacente persiste. Generalmente, esta resolución ocurre sin dejar secuelas permanentes como milia o cicatrices; si bien, es posible percibir áreas residuales de hipo o hiper-pigmentación.


