Was ist Morbus Fabry?
Morbus Fabry ist eine Erbkrankheit lysosomal Speicherstörung [1]. Es ist auch als Anderson-Fabry-Krankheit bekannt Angiokeratom Corporis diffusum.
Morbus Fabry verursacht Häufungen von Angiokeratome (kleine dunkelrote Flecken auf der Haut) und viele systemisch Symptome aufgrund Erklärung von Globotriaosylceramid (Gb3) in mehreren Organen.
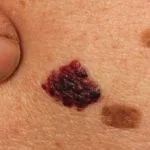
Angiokeratome bei Verdacht auf Fabry-Krankheit
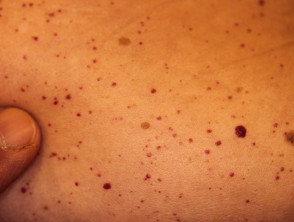
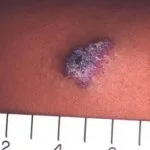
Angiokeratome bei Verdacht auf Fabry-Krankheit
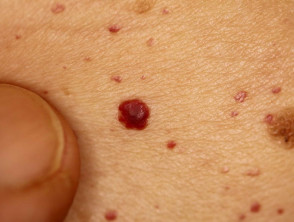
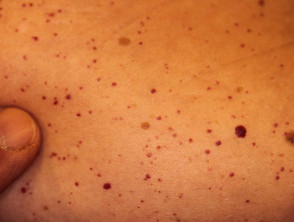
Angiokeratome bei Verdacht auf Fabry-Krankheit
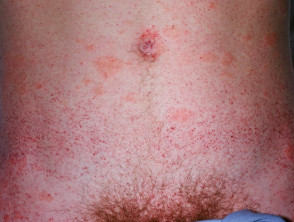
Angiokeratome bei Morbus Fabry
Wer erkrankt an Morbus Fabry?
Die Schätzung Vorherrschaft bei Männern beträgt sie etwa einen von 40.000 bis 60.000 Einwohnern [2]. Morbus Fabry betrifft im Allgemeinen Männer und Jungen schwerer und in einem jüngeren Alter als Frauen und Mädchen, da die Vererbung mit dem X-Chromosom (männlich) verbunden ist Sex Es hat nur ein X Chromosom während die Weibchen zwei haben). Morbus Fabry kann in allen ethnischen Gruppen auftreten. [3].
Das Erkrankungsalter des Morbus Fabry kann zwischen dem zweiten und dem fünften Lebensjahrzehnt oder später liegen.
Die Fabry-Krankheit weist oft unspezifische Symptome auf, die mild und subtil sein können, und wird häufig übersehen oder falsch diagnostiziert, was zu einer Unterschätzung ihrer Prävalenz führt. [4].
Was verursacht Morbus Fabry?
Morbus Fabry wird verursacht durch Mutation von Alpha-Galactosidase A Gen (GLA) auf dem langen Arm des X-Chromosoms abgebildet [5]. Es gibt Hunderte verschiedener Erreger Varianten der Mutation, die zu unterschiedlichen Symptomen und variablem Schweregrad führen [6]. Männer mit Morbus Fabry geben das Gen an alle ihre Töchter weiter, aber an keinen ihrer Söhne. Frauen mit Morbus Fabry haben eine 50%-Chance, das Gen an ihre Töchter oder Söhne weiterzugeben. Einige Frauen und Mädchen sind aufgrund von a genauso stark betroffen wie Männer und Jungen zufällig Inaktivierung des X-Chromosoms [7].
Gb3 ist ein Zwischenprodukt im Globosid-Abbauweg (ein Glykosphingolipid), ein Hauptbestandteil einiger Zellmembranen. Die Enzym Alpha-Galaktosidase A katalysiert die Ausschnitt (Abtrennung) der terminalen Galaktose von Gb3. Das Fehlen des Enzyms Alpha-Galaktosidase A führt zur Anreicherung von Gb3 in verschiedenen Geweben, was zum Zelltod führt. Die klinisch bedeutendsten Orte der Gb3-Akkumulation sind Blutgefäße der Haut, des Herzens, Nervenund Nieren [8].
Was sind die klinischen Merkmale des Morbus Fabry?
Die charakteristischen Merkmale des Morbus Fabry sind:
- Cluster von Angiokeratomen (klein, dunkelrot). Papeln)
- Hitzeunverträglichkeit und Hypohidrose (reduziertes Schwitzen)
- Episoden von Akroparästhesie (neuropathisch Schmerzen in Händen und Füßen (Akroparästhesie), Präzipitat aufgrund von Stress oder extremen Temperaturen
- Wolkendecke Hornhaut
- Hörverlust [9]
- Bauchbeschwerden wie Schmerzen, Übelkeit, Erbrechen und Durchfall oder Verstopfung [10]
- Wiederkehrend Fieber, Müdigkeit, psychische und soziale Probleme
- Schwerwiegende und lebensbedrohliche Komplikationen, einschließlich Nierenschäden, Herzerkrankungen und Schlaganfall [11].
Dermatologisch Demonstrationen treten bei mehr als 70% der Patienten mit Morbus Fabry auf, mit a halb Erkrankungsalter mit 17 Jahren [3].
Angiokeratome
Angiokeratome sind erweiterte Blutgefäße an der Oberseite Dermis, die sich als rote oder schwarze Papeln präsentieren. Die Papeln nicht weiß werden mit Druck und größer Verletzungen werden kann warzig. Cluster bzw diffus Angiokeratome, die erstmals bei jungen Erwachsenen auftreten, sollten Ärzte auf die mögliche Diagnose eines Morbus Fabry aufmerksam machen.
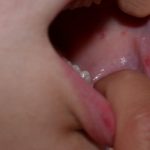
Bei der Fabry-Krankheit werden Angiokeratome durch die Ansammlung von Gb3 im Blut verursacht dermal endothelial Zellen, was zu Ausbeulungen und Insuffizienz der Gefäßwände führt [12]. Angiokeratome befinden sich meist im Badeanzugbereich (vom Nabel bis zum oberen Teil der Oberschenkel, einschließlich der Genitalien). Mehr als ein Drittel der Patienten auch entwickeln Angiokeratome und Teleangiektasie auf den Lippen und im Mund [13].
Es besteht kein Zusammenhang zwischen der Schwere der Erkrankung und dem Ausmaß der Angiokeratome. [14].
Hypohidrose
Hypohidrose ist ein häufiges Merkmal des Morbus Fabry und führt zu trockener Haut und Hitzeunverträglichkeit. [15]. Dies ist wahrscheinlich das Ergebnis einer Anomalie autonom Nerv Reaktion aufgrund von Gb3-Ablagerung.
Andere Haut- Manifestationen der Fabry-Krankheit
Morbus Fabry wird durch andere Hautzeichen erkannt, wie zum Beispiel:
- Lymphödem der Unterschenkel [14]
- Spärliche und dünne Gesichtsbehandlung Haar [14]
- Raynauds Phänomen [15].
Was sind die Komplikationen der Fabry-Krankheit?
Die drei schwerwiegendsten Komplikationen des Morbus Fabry sind Nierenerkrankungen, Herzerkrankungen und zerebrovaskulär Krankheit [3].
- Nierenerkrankungen reichen von isolierter Proteinurie bis zum Endstadium Nieren- Misserfolg [3].
- Herzerkrankungen, die zusammen mit Morbus Fabry auftreten, umfassen auch die linke Herzkammer Hypertrophie, Herzinsuffizienz, Herzinfarkt Arterie Krankheit und Arrhythmien.
- Zerebrovaskuläre Erkrankungen umfassen vorübergehend ischämisch ischämische Anfälle und Schlaganfälle.
Wie wird Morbus Fabry diagnostiziert?
Männliche und weibliche Patienten werden unterschiedlich diagnostiziert.
- Männliche Patienten können durch eine Blutuntersuchung diagnostiziert werden. Ein sehr niedriges Niveau der Alpha-Galactosidase-A-Aktivität (<3%) es suficiente para diagnosticar la enfermedad de Fabry, mientras que un nivel superior al 35% excluye la enfermedad de Fabry. Los niveles de actividad entre estas dos cifras requieren un genetisch Analyse zur Bestätigung der Diagnose.
- Bei weiblichen Patienten ist die Enzymaktivität unterschiedlich, wobei ein erheblicher Anteil eine normale Aktivität aufweist. Daher muss die Diagnose eines Morbus Fabry durch eine genetische Analyse gestellt werden.
- Wenn der Gentest keine krankheitsverursachende Mutation identifiziert, die für Morbus Fabry spezifisch ist, Biopsie der betroffenen Organe (einschließlich Hautbiopsie); das kann man zeigen intrazellulär Akkumulation von Gb3 [3,14].
Auch Angehörige des diagnostizierten Patienten sollten auf Morbus Fabry untersucht werden.
Urin Analyse und EKG sollte durchgeführt werden, um Nieren- und Herzerkrankungen zu erkennen Anomalien.
Welches ist das Differenzialdiagnose für Morbus Fabry?
Aufgrund seiner Seltenheit und der Vielzahl unspezifischer klinischer Merkmale wird die Fabry-Krankheit oft falsch diagnostiziert und umgangssprachlich als „große Betrügererkrankung“ angesehen. [16].
Angiokeratome sind nicht charakteristisch für Morbus Fabry, da bei gesunden Personen einzelne Läsionen auftreten können. Sie werden auch bei Patienten mit gefunden erblich hämorrhagisch Teleangiektasie und bei den anderen lysosomalen Erkrankungen, zu denen gehören:
- Schindler-Krankheit (eine seltene erbliche Stoffwechselstörung)
- Fukosidose (eine seltene lysosomale Speicherstörung, die eine lebensbedrohliche Ansammlung von Zucker verursacht)
- Aspartylglucosaminurie (eine Erkrankung, die auf einen Defekt des Enzyms Aspartylglucosaminidase zurückzuführen ist)
- Beta-Mannosidose (eine Erkrankung, die aus einer verminderten Aktivität des Enzyms Beta-Mannosidase resultiert)
- Sialidose (eine Stoffwechselstörung, die durch einen Mangel des Enzyms Neuraminidase verursacht wird) [12,14].
Die häufigsten Fehldiagnosen sind die folgenden:
- Rheumatologisch Bedingungen wie z rheumatisch Fieber oder Dermatomyositis – in diesen Fällen sind die dermatologischen Manifestationen unterschiedlich in Aussehen und zeitlichem Verlauf
- Neuropsychologische Erkrankungen und Fibromyalgie - bei denen keine Herz-, Nieren-, Nerven- und Hautbeteiligung vorliegt
- Polyzythämie wahr und wesentlich Thrombozythämie - Morbus Fabry verursacht keinen Anstieg Plättchen sagen
- Hereditäre hämorrhagische Teleangiektasie: Morbus Fabry verursacht normalerweise keine Nasenbluten oder Magen-Darm-Blutungen
- Reizdarm Syndrom - Dies hat keine anderen systemischen Manifestationen [4,17].
Was ist die Behandlung für Morbus Fabry?
Nach der Diagnose wird männlichen Patienten mit Morbus Fabry unabhängig von den klinischen Merkmalen häufig eine Enzymersatztherapie verschrieben. [9,14,18]. Weibliche Patienten sollten nur behandelt werden, wenn sie signifikante klinische Manifestationen aufweisen.
Die Enzymersatztherapie versorgt Patienten mit dem mangelhaften Enzym Alpha-Galactosidase A. Es reduziert die Schwere und das Fortschreiten von Krankheitserscheinungen, insbesondere neuropathischen Schmerzen und Nierenerkrankungen, indem es die Gewebeablagerung von Gb3 reduziert. Die Wirkung einer Enzymersatztherapie auf kardiovaskuläre Manifestationen ist weniger klar [18].
Patienten mit Morbus Fabry benötigen möglicherweise die Unterstützung verschiedener Fachrichtungen, darunter Hausärzte, Kardiologen, Nephrologen, Neurologen usw Dermatologen ihre individuellen organischen Manifestationen zu verwalten. Neuropathische Schmerzen an Händen und Füßen können eine Reaktion sein Antikonvulsiva, wie Carbamazepin [18,19].
Die Behandlung von Angiokeratomen umfasst Kryotherapie, Elektrokoagulation, Exzisionschirurgie und Sein Therapie [12,14,20].
Welche Folgen hat Morbus Fabry?
Morbus Fabry ist progressiv und führt im Allgemeinen zu einer kürzeren Lebenserwartung. Die Prognose variiert von Patient zu Patient [21]. Bei männlichen Patienten mit schweren Formen des Morbus Fabry ist die Überlebenszeit deutlich auf weniger als 60 Jahre, häufig auf 40 oder 50 Jahre, verkürzt. Die meisten Todesfälle sind auf Herz-Kreislauf-Erkrankungen zurückzuführen, darunter Schlaganfall, Herzerkrankungen, Herzinfarktund Herzinsuffizienz. Tod durch Nierenversagen, SeptikämieEs wurde auch über Selbstmorde berichtet [18]. Der Nutzen einer Enzymersatztherapie für die Gesamtprognose ist aufgrund des Mangels an aktueller Evidenz unklar [18,22,23].