Das sind mehrere endokrine Neoplasma Syndrome?
Multiple endokrine Neoplasie (MEN) -Syndrome sind meist Erbkrankheiten, bei denen verschiedene endokrine Erkrankungen auftreten Drüsen entwickeln gutartig (nicht krebsartig) oder der Böse Tumoren (krebsartig) oder Hyperplasie (Sie wachsen im Übermaß, ohne Tumore zu bilden).
Es gibt 2 Haupttypen von MÄNNERN, MÄNNER Typ 1 und MÄNNER Typ 2. MÄNNER Typ 2 ist in 3 Untertypen unterteilt:
- MÄNNER Typ 2A
- FMTC (Familie medulläre Schilddrüse Karzinom)
- MEN Typ 2B (früher bekannt als MEN Typ 3).
Multiple endokrine Neoplasie Typ 2B weist zusätzliche Merkmale auf, einschließlich Schleimhaut NeuromeNerv Tumoren in der schleimig Membranen), Neurome im Darm, die zu Magen-Darm-Störungen führen Anomalienund auffälliges Gesichtsaussehen in Verbindung mit marfanoider Gewohnheit (dünne, große, lange Finger und Zehen und gewölbter Gaumen wie bei der Marfan-Krankheit). Diese Funktionen können die ersten sein Zeichen eines Praktikanten Malignität und es sollte auf weitere Forschung drängen.
Was ist eine MEN Typ 2B Krankheit?
Wie die MEN-Typ-2A-Krankheit birgt auch die MEN-Typ-2B-Krankheit ein hohes Risiko Entwicklung medulläres Schilddrüsenkarzinom und Phäochromozytom (a vaskulär Tumor der Nebennieren Drüse was zu hohem Blutdruck führen kann).
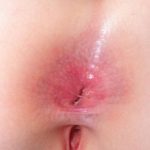
Die MEN-Typ-2B-Krankheit ist auch durch die Entwicklung multipler Neurome der Schleimhaut im frühen Leben gekennzeichnet. Neurome erscheinen als:
- Glänzende Beulen um Lippen, Zunge und Mundschleimhaut
- Klumpen auf den Augenlidern, die oft verdickt sind; Neurome können auch in der Hornhaut und Bindehaut
- Wucherungen im Magen-Darm-Trakt, die Verstopfung, Durchfall und in einigen Fällen einen vergrößerten Dickdarm (Megacolon) verursachen können.
Patienten mit MEN Typ 2B-Krankheit entwickeln häufig auch Anomalien der Wirbelsäule und Anomalien der Knochen der Füße und Oberschenkel. Viele haben lange Gliedmaßen und lockere Gelenke. Diese Eigenschaften sind zusammen mit verdickten Lippen und Augenlidern mit dem Marfanoid-Habitus (den Eigenschaften von Marfan) verbunden Syndrom).
Was ist die Ursache für die MEN Typ 2B Krankheit?
In 95%-Fällen ist die MEN-Typ-2B-Krankheit auf a zurückzuführen Mutation beim Tyrosinkinase RET-Domäne Gen am Codon 918 von Exon 16. Das RET-Gen ist ein Protokogen, was bedeutet, dass eine Mutation kann prädisponieren zur Bildung von Krebs.
Alle Subtypen von MEN 2 werden in a vererbt autosomal Die dominante Form und die Nachkommen der betroffenen Personen haben eine 50%-Chance, die Krankheit zu erben. Es werden jedoch nicht alle Fälle von MEN Typ 2B vererbt, da es einige Menschen mit dieser Krankheit gibt, die keine Familienanamnese haben. Es wurde festgestellt, dass ungefähr 50% bei betroffenen Personen die Mutation von einem Elternteil erbt und 50% eine neue Mutation aufweist.
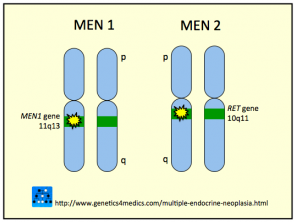
Genetik der multiplen endokrinen Neoplasie *
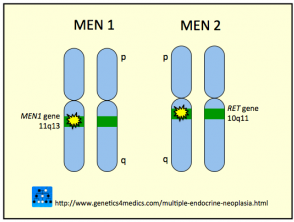
Multiple endokrine Neoplasie MEN
* Mit freundlicher Genehmigung von Genetics 4 Medics
Wie wurde die Diagnose gestellt?
Die Diagnose von MEN Typ 2B wird klinisch gestellt durch:
- Neurome der Schleimhäute der Lippen und der Zunge
- Marknervenfasern der Hornhaut im Auge
- unverwechselbar Fazies mit vergrößerten Lippen
- asthenischer Körper Habitus "Marfanoid"
- medulläres Schilddrüsenkarzinom
Molekular genetisch Die Tests können verwendet werden, um die Diagnose zu bestätigen, für Vorhersagetests und um vorgeburtlich Diagnose.
Was ist die Behandlung für MEN Typ 2B Krankheit?
Bei Menschen mit MEN Typ 2B sollte die Schilddrüse in einem sehr jungen Alter (ca. 1 Jahr) entfernt werden, um das Risiko einer Schilddrüsenerkrankung zu verringern Krebs. Im Vergleich zu anderen Arten von Schilddrüsenkrebs ist das medulläre Schilddrüsenkarzinom ein sehr aggressiver Krebs. Wenn die Schilddrüse im Kindesalter nicht entfernt wird, liegt das durchschnittliche Todesalter bei Menschen mit MEN Typ 2B-Krankheit bei etwa 21 Jahren. Nachdem die Schilddrüse entfernt wurde, müssen die Patienten für den Rest ihres Lebens Schilddrüsenhormonersatz (Thyroxin) einnehmen.
Es ist keine Heilung für eines der MEN-Syndrome bekannt. Patienten werden wegen ihrer Symptome behandelt. Für alle Patienten wird ein jährliches biochemisches Urin-Screening auf Phäochromozytom empfohlen. Gentests sind auch für Menschen mit einer Familiengeschichte der Krankheit wichtig, da eine frühzeitige Diagnose und Behandlung das Risiko für Schilddrüsenkrebs und Tod verringert.