Entendiendo los Síndromes de Neoplasia Endocrina Múltiple (MEN)
Definición y Tipos de Síndromes de Neoplasia Endocrina Múltiple
Los Síndromes de Neoplasia Endocrina Múltiple (MEN) constituyen mayormente afecciones hereditarias. En estos síndromes, varias glándulas endocrinas tienden a desarrollar tumores clasificados como benignos (no cancerosos) o malignos (cancerosos), o bien pueden presentar hiperplasia, que es el crecimiento excesivo sin la formación de tumores definidos.
Existen dos clasificaciones principales de MEN: el MEN tipo 1 y el MEN tipo 2. El MEN tipo 2 se subdivide a su vez en tres subtipos distintos:
- MEN tipo 2A
- Carcinoma Medular de Tiroides Familiar (FMTC, por sus siglas en inglés, familiar tiroides medular carcinoma)
- MEN tipo 2B (anteriormente conocido como MEN tipo 3)
El síndrome de Neoplasia Endocrina Múltiple tipo 2B presenta características clínicas adicionales distintivas. Estas incluyen neuromas de la mucosa (nervio tumores localizados en las membranas mucosas), neuromas en el tracto intestinal que derivan en anormalidades gastrointestinales, y un fenotipo facial característico asociado al hábito marfanoide (como dedos largos y delgados, complexión alta y paladar arqueado, similar al síndrome de Marfan Comprendiendo el Síndrome de Marfan: Causas y Características Principales ¿Qué es exactamente el Síndrome de Marfan? El síndrome de Marfan es un trastorno genético que afecta el tejido conectivo, el cual es fundamental para unir y dar soporte estructural al organismo. Este trastorno impacta predominantemente tres sistemas corporales: el sistema esquelético (huesos y articulaciones), el cardiovascular (corazón y vasos sanguíneos) y los ojos. Las manifestaciones clínicas más notables del síndrome más). La identificación temprana de estos signos es crucial, ya que pueden ser la primera señal de una malignidad subyacente y ameritan una investigación médica exhaustiva.
Comprendiendo el Síndrome de Marfan: Causas y Características Principales ¿Qué es exactamente el Síndrome de Marfan? El síndrome de Marfan es un trastorno genético que afecta el tejido conectivo, el cual es fundamental para unir y dar soporte estructural al organismo. Este trastorno impacta predominantemente tres sistemas corporales: el sistema esquelético (huesos y articulaciones), el cardiovascular (corazón y vasos sanguíneos) y los ojos. Las manifestaciones clínicas más notables del síndrome más). La identificación temprana de estos signos es crucial, ya que pueden ser la primera señal de una malignidad subyacente y ameritan una investigación médica exhaustiva.
Características Clínicas de la Enfermedad MEN tipo 2B
Al igual que sucede con el MEN tipo 2A, la enfermedad MEN tipo 2B conlleva un riesgo elevado de desarrollo de carcinoma medular de tiroides y feocromocitomaEntendiendo el Feocromocitoma: Definición, Prevalencia y Causas Genéticas El feocromocitoma (a veces escrito feocromocitoma en la nomenclatura estadounidense) es un trastorno neuroendocrino poco común caracterizado por un tumor que produce y secreta cantidades excesivas de catecolaminas, principalmente noradrenalina y, en menor grado, adrenalina. Estos tumores tienen su origen primario en la médula suprarrenal (aproximadamente el 85% de los casos). Los casos restantes (15%) se desarrollan a partir de neural ganglios más. Este último es un tumor vascular localizado en la suprarrenal glándula, capaz de provocar hipertensión arterial.
El rasgo definitorio del MEN tipo 2B es la aparición temprana de múltiples neuromas de la mucosa. Estos neuromas se manifiestan típicamente como:
- Bultos brillantes localizados alrededor de los labios, la lengua y el revestimiento bucal.
- Nódulos en los párpados, que a menudo resultan engrosados; los neuromas también pueden observarse en la córnea y la conjuntiva.
- Crecimientos dentro del tracto gastrointestinal que pueden manifestarse como estreñimiento, diarrea e, incluso, provocar la aparición de megacolon (agrandamiento del colon).
Los pacientes afectados por MEN tipo 2B frecuentemente presentan anomalías esqueléticas en la columna vertebral, los huesos de los pies y los muslos. Es común que exhiban extremidades largas y laxitud articular. Estas conformaciones físicas, aunadas a los labios y párpados de apariencia engrosada, componen el hábito marfanoide (o características propias del síndrome de Marfan).
Causas Genéticas de la Enfermedad MEN tipo 2B
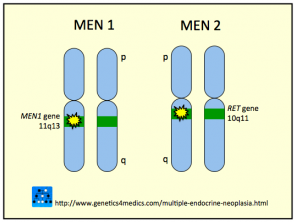
En la gran mayoría de los casos (95%), la enfermedad MEN tipo 2B se origina debido a una mutación específica en el dominio de tirosina quinasa del gen RET, concretamente en el codón 918 del exón 16. El gen RET es considerado un protooncogén, lo que significa que una alteración genética en su estructura puede predisponer al organismo a desarrollar neoplasias malignas.
Es importante destacar que todos los subtipos de MEN comparten una base genética común que involucra alteraciones en el gen RET.
La herencia de la neoplasia endocrina múltiple tipo 2B (MEN 2B) sigue un patrón autosómico dominante. Esto significa que la descendencia de un individuo afectado tiene un 50% de probabilidad de heredar la enfermedad. No obstante, es importante destacar que no todos los casos de MEN 2B son hereditarios; aproximadamente el 50% de los individuos con la afección no presentan antecedentes familiares. Se ha determinado que de ese 50% no hereditario, la mitad corresponde a una nueva mutación genética aparecida en el paciente.
Genética de la Neoplasia Endocrina Múltiple
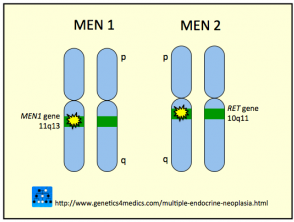
Neoplasia endocrina múltiple MEN
* Imagen cortesía de Genetics 4 Medics
Diagnóstico Clínico de la Enfermedad MEN Tipo 2B
El diagnóstico de MEN tipo 2B se establece clínicamente mediante la identificación de las siguientes características:
- Neuromas de las mucosas presentes en labios y lengua.
- Fibras nerviosas corneales meduladas observables en el ojo.
- Un facies característico con labios notablemente agrandados.
- Un biotipo corporal asténico, a menudo descrito como "marfanoide".
- La presencia de carcinoma medular de tiroides.
Además del examen clínico, las pruebas moleculares o genéticas son herramientas cruciales para confirmar el diagnóstico, realizar pruebas predictivas en familiares y llevar a cabo el diagnóstico prenatal si es necesario.
Tratamiento y Manejo del Síndrome MEN Tipo 2B
Para las personas diagnosticadas con la enfermedad MEN tipo 2B, la intervención más crítica es la tiroidectomía (extirpación quirúrgica de la glándula tiroides) a una edad muy temprana, idealmente alrededor del primer año de vida. Esta medida agresiva es fundamental para mitigar el riesgo extremadamente alto de desarrollar cáncer de tiroides. El carcinoma medular de tiroides asociado a MEN 2B es notablemente agresivo; si la tiroides no se extirpa durante la infancia, la esperanza de vida promedio para estos pacientes desciende drásticamente, situándose alrededor de los 21 años.
Tras la tiroidectomía, los pacientes requieren terapia de reemplazo hormonal con tiroxina por el resto de sus vidas para mantener las funciones metabólicas normales. Actualmente no existe una cura definitiva para ninguno de los síndromes MEN; por lo tanto, el manejo se centra en el tratamiento sintomático y la vigilancia continua.
Se recomienda encarecidamente realizar una detección bioquímica anual de feocromocitoma en todos los pacientes con MEN 2B. Asimismo, la realización de pruebas genéticas es fundamental para los individuos con antecedentes familiares conocidos, ya que el diagnóstico precoz y el tratamiento oportuno son las estrategias más efectivas para reducir significativamente el riesgo de cáncer de tiroides y mortalidad asociada a la condición.


