Was ist Crouzon? Syndrom?
Das Crouzon-Syndrom ist durch eine Vielzahl von kraniofazialen Erkrankungen gekennzeichnet Entwicklung Symptome.
Ist ein erblich geerbter Zustand in a autosomal dominantes Muster (ein abnormales Gen eines Elternteils kann das Syndrom verursachen). Es ist auch bekannt als Crouzon-Krankheit, kraniofaziale Dysostose, Kraniostenose, Apert-Crouzon-Syndrom, Typ-II-Akrozephalosyndaktylie, Vogt-Cephalosyndaktylie und Trigorrindaktylie. Dysplasie.
Crouzon beschrieb es erstmals 1912.
Wer bekommt das Crouzon-Syndrom?
Das Crouzon-Syndrom ist selten, obwohl es nach wie vor das häufigste Craniosynostose-Syndrom ist (bei dem sich die fibrösen Schädelgelenke im Kindesalter vorzeitig schließen).
- Es betrifft 1 von 60.000 Lebendgeburten.
- Es scheint bei Menschen aller Rassen und Ethnien gleichermaßen diagnostiziert zu werden.
- Es wird häufig bei der Geburt oder im Säuglingsalter aufgrund charakteristischer Gesichtszüge diagnostiziert.
Was verursacht das Crouzon-Syndrom?
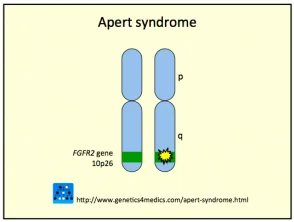
Das Crouzon-Syndrom wird normalerweise durch verursacht Mutationen beim Fibroblasten Wachstumsfaktor Empfänger 2 (FGFR2) gen. das FGFR3 Das Gen kann auch beteiligt sein.
- Diese Mutation führt dazu, dass unreife Zellen während der Embryogenese zu Knochenzellen werden.
- In 50%-Fällen liegt eine Familienanamnese des Crouzon-Syndroms vor.
- In den anderen 50%-Fällen ist das Syndrom aufgrund neuer genetischer Mutationen sporadisch.
Genetik des Crouzon-Syndroms *
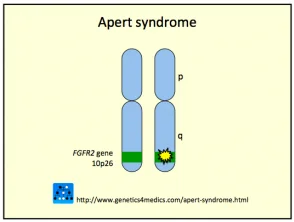
Crouzon2-Syndrom
* Mit freundlicher Genehmigung von Genetics 4 Medics
Was sind die klinischen Merkmale des Crouzon-Syndroms?
Die klinischen Merkmale des Crouzon-Syndroms variieren stark und reichen von leicht bis schwer. Das Hauptmerkmal ist das vorzeitige Schließen der Nähte der Kappe und der Schädelbasis sowie die Kraniosynostose.
Gesichtszüge
Verwandte charakteristische Gesichtsmerkmale sind:
- Exophthalmus (abnormaler Vorsprung des Augapfels)
- Hypertelorismus (übermäßige Breite zwischen den Augen)
- Hypoplastischer Oberkiefer (ein unterentwickelter Kiefer)
- Unterkieferprognathie (Vorsprung des Unterkiefers)
- Eine kurze Oberlippe
- Spitze Nase.
Sehstörungen
Zu den mit dem Crouzon-Syndrom verbundenen Sehstörungen gehören:
- Amblyopie (dunkles Sehen ohne erkennbare Veränderungen der Augenstrukturen)
- Ametropiefeuerfest Error)
- Strabismus (Unfähigkeit zu erreichen Fernglas Sehvermögen aufgrund von Muskelungleichgewichten in den Augäpfeln).
Diese Sehstörungen sind auf eine Hornhautverletzung zurückzuführen. Wasserfälle (eine Trübung der Augenlinse oder der transparenten Membran, die sie umgibt), optisch Atrophie (Verschlechterung der Optik Nerv) und Iriskolobom (ein Loch in der Iris).
Andere Eigenschaften
Weitere mit dem Crouzon-Syndrom verbundene Merkmale sind:
- Verminderte geistige Funktion und erhöhtes Risiko. intrakraniell Druck und Krämpfe
- Atemwegsbeschwerden durch Verengung der Nasopharyngeal Passage und abweichend Fruchtfleisch oder andere strukturelle Anomalien
- Hörverlust und / oder Ménière Krankheit (eine Störung des Innenohrs, die durch Episoden von gekennzeichnet ist Schwindel, Klingeln in den Ohren, Hörverlust und Druck im Ohr)
- Skelettanomalien, einschließlich Fusion der Wirbelsäule.
Die wichtigsten dermatologischen Zeichen Crouzon-Syndrom ist Akanthose Nigricans, bei denen es zu einer Verdickung kommt, hyperpigmentiert Haut mit einem samtigen Gefühl, das Hals, Rumpf und Gesicht betrifft. Es tritt normalerweise zu Beginn der Pubertät auf.
Acanthosis nigricans *
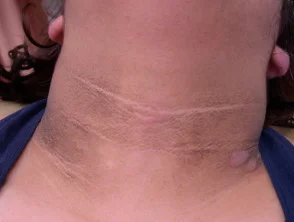
Acanthosis nigricans
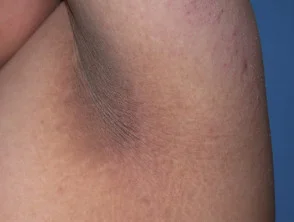
Acanthosis nigricans
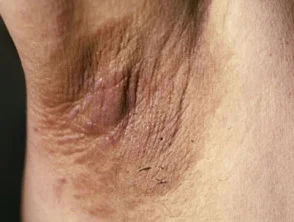
Acanthosis nigricans
Wie wird das Crouzon-Syndrom diagnostiziert?
Crouzon wird bei einem Kind mit Craniosynostose sicher diagnostiziert, wenn Mutationen in der FGFR2 Gene werden nachgewiesen. das Gene der Eltern des Betroffenen kann auch auf Mosaik untersucht werden (mit Zellen von zwei oder mehr genetisch unterschiedlichen Typen).
Wie wird das Crouzon-Syndrom behandelt?
Die Standardbehandlung für das Crouzon-Syndrom umfasst eine frühe Kraniektomie (die chirurgische Entfernung eines Teils des Schädels) und eine kosmetische Rekonstruktion, um das normale Gesichtswachstum zu fördern.
Multidisziplinär Die Pflege kann die medizinische und chirurgische Beurteilung und Behandlung von Symptomen umfassen. Dies kann Folgendes umfassen:
- Ophthalmologisch Behandlung von Amblyopie, Ametropie und Strabismus
- Audiologische Behandlung und Myringotomie bei Hörverlust
- Shunt zur Behandlung des Hirndrucks aufgrund von Hydrozephalus (erhöhte Liquor cerebrospinalis in der Schädelhöhle)
- Tracheotomie (a Einschnitt in der Luftröhre) zur Behandlung von Atemwegsobstruktion
- Kieferorthopädische Chirurgie zur Behandlung von Unregelmäßigkeiten in Zähnen und Kiefern.
Ausblick Wie ist der Ausblick für das Crouzon-Syndrom?
Verbesserungen der chirurgischen Techniken, insbesondere der kraniofazialen Chirurgie, haben die Lebensqualität, die intellektuellen und körperlichen Fähigkeiten und die soziale Akzeptanz von Kindern mit Crouzon-Syndrom erheblich verbessert.
Die Lebenserwartung von Menschen mit Crouzon-Syndrom ist in der Regel normal, aber Mortalität kann aufgrund einer Atemwegsobstruktion auftreten, akut Kurzatmigkeit oder erhöhter Hirndruck.


