¿Qué es el Síndrome de Crouzon?
El síndrome de Crouzon se define por un conjunto diverso de afecciones craneofaciales y di sviluppo que presentan diversas sintomatologías.
Esta es una condición hereditaria transmitida en un patrón autosomico dominante; significa que un gene anormal heredado de cualquiera de los padres puede desencadenar el síndrome. También se le conoce por varios sinónimos, incluyendo enfermedad de Crouzon, disostosis craneofacial, craneostenosis, síndrome de Apert-Crouzon, acrocefalosindactilia tipo II, cefalosindactilia de Vogt y displasia trigorrindactílica.
El médico que describió esta condición por primera vez fue Crouzon, en el año 1912.
Prevalencia del Síndrome de Crouzon
El síndrome de Crouzon es clasificado como poco común, aunque representa el síndrome de craneosinostosis más frecuente (condición donde las suturas fibrosas del cráneo se fusionan de manera prematura durante la infancia).
- Afecta aproximadamente a 1 de cada 60,000 recién nacidos vivos.
- No se observan diferencias significativas en el diagnóstico entre personas de distintas razas y etnias.
- Frecuentemente se diagnostica al momento del nacimiento o durante la infancia debido a sus rasgos faciales característicos.
Causas Genéticas del Síndrome de Crouzon
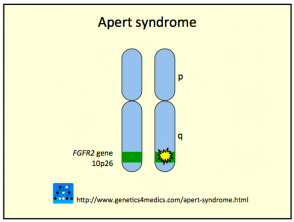
El síndrome de Crouzon es provocado generalmente por mutazioni en el gen del fibroblastico fattore di crescita recettore del 2 (FGFR2). En algunos casos, el gen FGFR3 también puede estar implicado en su origen.
- Questa mutazione induce señales en las células inmaduras para que se diferencien prematuramente en células óseas durante la etapa de embriogénesis.
- En el 50% de los casos diagnosticados, existe un historial familiar documentado del síndrome de Crouzon.
- El otro 50% de los casos son esporádicos, originados por nuevas y espontáneas mutaciones genéticas.
Genética del Síndrome de Crouzon *
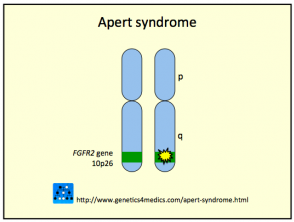
Síndrome de Crouzon2
* Immagine per gentile concessione di Genetics 4 Medics
Características Clínicas del Síndrome de Crouzon
Las manifestaciones clínicas del síndrome de Crouzon presentan una gran variabilidad, abarcando desde formas leves hasta muy severas. El signo distintivo fundamental es la craneosinostosis, que implica el cierre prematuro de las suturas del cráneo y de la base craneal.
Rasgos Faciales Distintivos
Las particularidades faciales frecuentemente observadas incluyen:
- Exoftalmos (una proyección anormal y notable de los globos oculares).
- Hipertelorismo (un aumento en la distancia entre los ojos).
- Hipoplasia maxilar (un desarrollo insuficiente o poco desarrollado de la mandíbula superior).
- Prognatismo mandibular (un avance o protrusión de la mandíbula inferior).
- Un labio superior notablemente corto.
- Nariz con una forma puntiaguda.
Defectos Oculares Asociados
Entre las afecciones visuales que pueden acompañar al síndrome de Crouzon se encuentran:
- Ambliopía (conocida como "ojo vago", que resulta en una visión pobre sin causas aparentes de daño físico).
Comprender las causas genéticas y reconocer las características clínicas tempranas son cruciales para el manejo efectivo del síndrome de Crouzon y sus complicaciones asociadas.
- Ametropía (error refractivo)
- Estrabismo (incapacidad para lograr visión binocular debido a desequilibrios musculares en los globos oculares).
Estos defectos visuales pueden ser consecuencia de una lesión corneal, la presencia de cataratas (una opacidad del cristalino ocular o de la membrana transparente que lo recubre), atrofia óptica (deterioro del nervio óptico) y coloboma del iris (una abertura o defecto en el iris).
Otras Características Clínicas del Síndrome de Crouzon
El síndrome de Crouzon se asocia con diversas características adicionales, entre ellas:
- Disminución de la función mental y un riesgo elevado de presión intracraneal y convulsiones.
- Manifestaciones respiratorias derivadas del estrechamiento del pasaje nasofaríngeo y alteraciones estructurales como una pulpa desviada u otras anormalidades.
- Pérdida auditiva y/o la enfermedad de Ménière (un trastorno del oído interno caracterizado por episodios de vértigo, tinnitus, hipoacusia y sensación de presión en el oído).
- Anomalías esqueléticas, como la fusión de las vértebras espinales.
El signo dermatológico predominante en el síndrome de Crouzon es la acantosis nigricans. Esta condición se manifiesta con engrosamiento, hiperpigmentación y una textura aterciopelada de la piel, afectando típicamente el cuello, el torso y la cara. Generalmente, este síntoma emerge al comienzo de la pubertad.
Manifestaciones de Acantosis Nigricans *
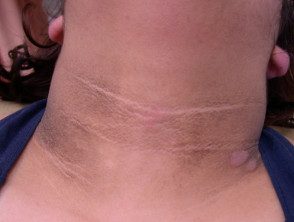
Acantosi nigricans
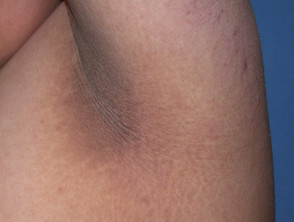
Acantosi nigricans
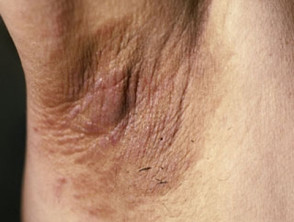
Acantosi nigricans
Diagnóstico del Síndrome de Crouzon
El diagnóstico del síndrome de Crouzon se establece con certeza en un niño que presenta craneosinostosis mediante la detección de mutaciones en los genes FGFR2. Adicionalmente, es posible realizar evaluaciones genéticas a los padres del individuo afectado para identificar la presencia de mosaicismo (la coexistencia de dos o más tipos de células genéticamente distintas en el organismo).
```html


