Was ist Legius? Syndrom?
Das Legius-Syndrom ist selten genetisch Störung, die erstmals 2007 beschrieben wurde [1]. Auch bekannt als Neurofibromatose Typ-1-Syndrom [2].
Das Legius-Syndrom ist klassisch durch mehrere gekennzeichnet Macules, bekannt als Café au lait Macules [3]. Im Gegensatz zur Typ-1-Neurofibromatose werden beim Legius-Syndrom keine Tumoren gefunden.
Kaffee mit Milchmakula
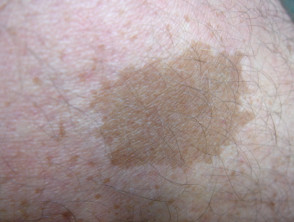
Café au lait macule
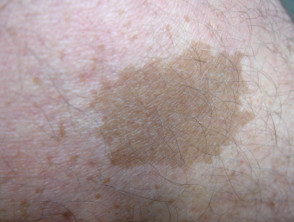
Café au lait macule
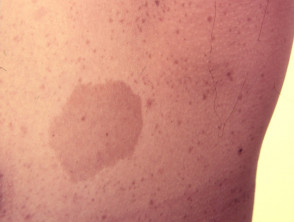
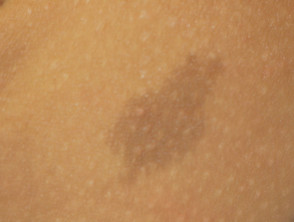
Café au lait macule
Wer bekommt das Legius-Syndrom?
Das Legius-Syndrom kann bei jedem auftreten, der mindestens einen leiblichen Elternteil mit genetisch bestätigtem Legius-Syndrom hat [2]. das Vorherrschaft Das Legius-Syndrom ist unbekannt, kann jedoch aufgrund einer Fehldiagnose der Typ-1-Neurofibromatose höher sein als erwartet [4].
Was verursacht das Legius-Syndrom?
Das Legius-Syndrom ist eine genetische Erkrankung, die in a vererbt wird autosomal dominante Art und Weise mit a SPRING1 Gen Mutation im Chromosom 15q14. Diese Mutation führt zu einer abnormalen Funktion von SPRED1 Protein, die für die Regulierung der spezifischen Signalwege der Zellen verantwortlich ist Proliferation, Differenzierungund Apoptose. Kein anderer Erreger Es wurde festgestellt, dass die Varianten das Legius-Syndrom verursachen. [2,3].
Als ein autosomal dominant Bedingung, Legius-Syndrom kann bei jedem mit mindestens einem leiblichen Elternteil mit genetisch bestätigtem Legius-Syndrom auftreten. In seltenen Fällen kann es auch vorkommen de novoDiese Fälle wurden jedoch nicht ausreichend bewertet, um dies zu bestätigen [2]. Jedes Kind mit einem Elternteil mit genetisch bestätigtem Legius-Syndrom hat ein 50%-Risiko, die Krankheit zu erben.
Was sind die klinischen Merkmale des Legius-Syndroms?
Die klinischen Merkmale des Legius-Syndroms unterscheiden sich in Art und Schweregrad von Person zu Person erheblich.
Fast alle Patienten mit Legius-Syndrom haben mehrere Café-au-lait-Makula [2,3]. Die Anzahl dieser Makula nimmt im Kindesalter tendenziell zu. [4].
Andere häufig Haut- Funktionen können umfassen:
- Sommersprossen in den Achseln und Leisten Regionen
- Lipome [2–4].
Nicht-kutane Merkmale können sein:
- Makrozephalie
- Ungewöhnliche Gesichtszüge, ähnlich dem Noonan-Syndrom
- Kleinwuchs
- Pectus excavatum (versunkenes Sternum) oder Pectus carinatum (hervorstehendes Sternum) [2–4].
Neuropsychiatrische Merkmale können umfassen:
- Lernschwierigkeiten
- Entwicklung verzögern
- Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) [2–4].
Intellektuelle Behinderungen sind bei Patienten mit Legius-Syndrom im Allgemeinen weniger schwerwiegend als bei Patienten mit Typ-1-Neurofibromatose [5].
Wichtig ist, dass das Legius-Syndrom keine Ursachen hat Neurofibrome und Lisch Knötchen, die typischerweise bei Typ-1-Neurofibromatose auftreten [2,3].
Wie wird das Legius-Syndrom diagnostiziert?
Das Legius-Syndrom ist allein aufgrund der klinischen Symptome schwer zu diagnostizieren, da sein kutanes klinisches Erscheinungsbild ähnlich wie bei anderen Erkrankungen mit mehreren Café-du-lait-Makula ist.
Bei Verdacht auf ein Legius-Syndrom können Gentests verwendet werden, um die Diagnose zu bestätigen. Das beinhaltet:
- Sequenzanalyse SPRING1
- Deletions- / Duplikationsanalyse (wenn die Sequenzanalyse nicht bemerkenswert ist)
- Ein Panel mit mehreren Genen für SPRING1 und ein anderer Gene von Interesse [2,3].
Zu den verdächtigen Befunden, die Gentests für das Legius-Syndrom rechtfertigen könnten, gehören:
- Café au lait Macules ohne andere klinische Merkmale, was auf eine Typ-1-Neurofibromatose hindeutet
- Ein Vater mit Café au lait Macules ohne klinische Merkmale, was wiederum auf eine Typ-1-Neurofibromatose hinweist [2].
Welches ist das Differenzialdiagnose für Legius-Syndrom?
Das Legius-Syndrom wird oft falsch diagnostiziert, weil es Pigment Demonstrationen sind denen bei anderen Syndromen mit multiplen sehr ähnlich Lentigos. Eine korrekte Diagnose ist wichtig, um die langfristige Kontrolle und Verwaltung zu steuern.
Neurofibromatose Typ 1
- Die Typ-1-Neurofibromatose ist aufgrund ihrer ähnlichen klinischen diagnostischen Kriterien die häufigste Fehldiagnose [2–4].
- Neurofibrome, Lisch-Knoten, Knochen Anomalienund optische Gliome treten beim Legius-Syndrom nicht auf.
- Diese beiden Zustände sind bei jüngeren Kindern schwer zu unterscheiden, da dies bei den bei Neurofibromatose beobachteten charakteristischen Tumoren im Allgemeinen nicht der Fall ist. entwickeln bis später im Leben.
Noonan-Syndrom mit mehreren Lentiginen
- Das Noonan-Syndrom mit multiplen Lentigos (früher als LEOPARD-Syndrom bekannt) umfasst Herz-Kreislauf-, Ohr- und Genitalanomalien, die beim Legius-Syndrom nicht auftreten) [6].
Andere Syndrome, die durch mehrere Lentigos gekennzeichnet sind, umfassen:
- Autosomal dominant Latte Macules
- McCune-Albright-Syndrom.
Was ist die Behandlung für das Legius-Syndrom?
Kinder mit Legius-Syndrom sollten regelmäßig untersucht und evaluiert werden, um Entwicklungsverzögerungen, kognitiven Verfall und Verhaltensprobleme festzustellen. [2].
Die Behandlung des Legius-Syndroms ist in erster Linie unterstützend und sollte sich auf die spezifischen Probleme des Betroffenen konzentrieren.
Eine angemessene Verwaltung kann, falls angegeben, Folgendes umfassen:
- Körper-, Sprach- und / oder Ergotherapie
- Verhaltensmodifikationstherapie und medikamentöse Therapie (dh bei ADHS) [2].
Was ist das Ergebnis des Legius-Syndroms?
Patienten mit Legius-Syndrom haben eine gute Prognose mit richtigem problembasierten Management.
Eine betroffene Person muss das 50%-Risiko einer genetischen Übertragung auf jedes Kind berücksichtigen.