Was ist Hyperimmunoglobulinämie D mit Perioden Fieber Syndrom?
Hyperimmunoglobulinämie D mit periodischem Fiebersyndrom (MIM 260920) ist häufiger als Hyper-IgD-Syndrom oder HIDS bekannt. Es ist eine seltene geerbte autoinflammatorisch Syndrom, das mit präsentiert wiederkehrend Episoden von Fieber, Haut Eruption, Bauchschmerzen, Kopfschmerzen und Vergrößerung Lymphe Drüsen das beginnt in der Kindheit.
Mevalonazidurie ist eine schwere Variante von HIDS.
Wer bekommt HIDS und warum?
HIDS wird überwiegend in Populationen aus zwei Gebieten Nordeuropas, den Niederlanden (mehr als 50% Fälle) und Nordfrankreich identifiziert. Es wurden jedoch ungefähr 200 Fälle in verschiedenen ethnischen Gruppen gemeldet.
Die Symptome von HIDS beginnen normalerweise vor einem Jahr mit dem Median Das Erkrankungsalter beträgt 6 Monate. Die Diagnose zu stellen ist jedoch schwierig und kann oft viele Jahre dauern.
HIDS wird als geerbt autosomal rezessiver Zustand, dh zwei Kopien des Defekts Gen werden geerbt, einer von jedem nicht betroffen Träger Eltern.
Molekular Biologie und Genetik
Das an HIDS beteiligte MVK-Gen befindet sich in Chromosom 12 (12q24) und erzeugt die Enzym Mevalonatkinase. Mutationen in diesem Gen wurden in der 75% von typischen Fällen berichtet. V377I ist die häufigste krankheitsbedingte Erkrankung Mutation berichtet. Bei HIDS führen genetische Mutationen zu einer verringerten Enzymaktivität, etwa 5-10% normal. Mevalonazidurie ist eine schwerwiegendere und tödlichere Form dieses Mangels, bei der die Enzymaktivität praktisch nicht nachweisbar ist. (Schau runter)
Das Enzym Mevalonatkinase ist am Isoprenoidweg von beteiligt Cholesterin Biosynthese. Ein Enzymmangel führt zu einer Anreicherung von Mevalonsäure und einer Zunahme Interleukin 1.
HIDS klinische Merkmale
das akut fieberhaft HIDS-Episoden beginnen in der Kindheit. Die klinischen Merkmale sind:
- Fieber
- nicht wandernd makulopapulär Akne
- Kopfschmerzen
- schmerzhaft vergrößert Lymphknoten im Nacken
- Bauch- und Gelenkschmerzen.
Die Angriffe dauern bis zu einer Woche und sind länger als die von Familie Mittelmeerfieber. Welches kann wiederholen alle 1-2 Monate.
Die Eigenschaften wiederkehrender akuter Episoden von HIDS sind in der folgenden Tabelle beschrieben.
| Symptom | Eigenschaften |
|---|---|
| Fieber |
|
| Akne |
|
| Kopfschmerzen |
|
| Vergrößerte Lymphknoten im Nacken |
|
| Bauchschmerzen |
|
| Gelenkschmerzen |
|
| Vergrößerte Leber und Milz (Hepatosplenomegalie) |
|
| Tendinitis |
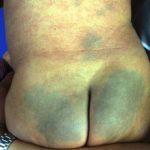
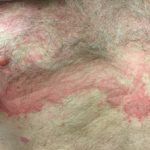
Haut Anzeichen von HIDS
Der Ausschlag betrifft bis zu 80% bei Patienten. Verschiedene Skins Eruptionen oder Eruptionen wurden bei diesem Syndrom beschrieben, und diese klingen langsam ab, sobald sich die fieberhafte Episode gelegt hat.
Die bei HIDS beobachteten Hautausschläge werden am häufigsten wie folgt beschrieben:
- kleine flache StellenMacules)
- erhabene Beulen (klein Papeln oder älter Knötchen)
- Masernartiger Ausschlagmorbilliform)
- bienenstockartiger AusschlagUrtikaria).
Weniger häufige oder seltene Hautpräsentationen umfassen:
- Henoch-Schönlein lila
- Erythem Elevatum Diutinum
- Petechien (kleine Blutungen oder violette Flecken)
- knotiges Erythem.
Mündlich und / oder vaginal aphthös Geschwüre beeinflussen 50% bei Patienten.
Was löst die Angriffe aus?
Akute Episoden können ausgelöst werden durch:
- Impfstoffe: Mehr als 50% berichten über mindestens eine Kindheitsepisode nach der Impfung
- Infektion
- Körperlicher und emotionaler Stress
- Trauma, einschließlich Operation
Was ist das wahrscheinliche Ergebnis für Patienten?
Es gibt eine Tendenz, sich mit dem Alter zu verbessern, mit weniger häufigen und weniger schweren Anfällen im Erwachsenenalter. Zwischen den Episoden ist die Gesundheit normal.
Eine kleine Untergruppe betroffener Patienten entwickeln neurologisch Anomalien im Erwachsenenalter ähnlich wie Mevalonazidurie (siehe unten).
Im Gegensatz zum bekannten Mittelmeerfieber Amyloidose selten bei HIDS gesehen, die weniger als 3% betreffen.
Die Lebenserwartung ist normalerweise normal, kann jedoch durch beeinflusst werden Nieren- Versagen durch Amyloidose oder schwer Infektionen.
Wie wird HIDS diagnostiziert?
Die charakteristischen wiederkehrenden akuten fieberhaften Anfälle ohne eindeutige Infektion oder Autoimmun Ursache, legt die Notwendigkeit einer Untersuchung nahe.
Klinische Kriterien
Zusätzlich zu den oben beschriebenen fieberhaften Anfällen sollten die klinischen diagnostischen Kriterien Folgendes umfassen:
- Beginn vor dem 5. Lebensjahr
- Die Folgen dauern weniger als 14 Tage
MVK-Genmutationen sind unwahrscheinlich, wenn diese Merkmale nicht vorhanden sind.
IgD-Spiegel
Erhöhte IgD-Spiegel treten bei vielen, aber nicht allen Patienten auf, insbesondere bei Kindern unter 3 Jahren. Die Level steigen nicht nur während eines Angriffs, sondern auch zwischen den Angriffen. Erhöhungen der IgD-Spiegel können bei anderen periodischen fieberhaften Syndromen wie familiärem Mittelmeerfieber und TRAPS und anderen auftreten chronisch entzündlich Bedingungen, so sollte es mit Vorsicht interpretiert werden.
Andere nützliche Tests
Organische Säuren im Urin, die während eines akuten Anfalls gemessen werden, weisen normalerweise erhöhte Mevalonsäurespiegel auf.
Während eines Angriffs steigt auch die Anzahl der weißen Blutkörperchen (Leukozytose), Erythrozyten Sedimentationsrate (ESR), C-reaktiv Protein (CRP) und Serum Amyloid A (SAA). Serum-IgA-Spiegel können ebenfalls ansteigen.
Radiometrische Assay-Tests können eine verringerte Mevalonatkinase-Aktivität in weißen Blutkörperchen oder in Kulturen nachweisen Fibroblasten.
Haut Biopsie kann eine leichte zeigen Vaskulitis das kann tief gehen. Die Veränderungen können Sweet-Krankheit oder Cellulite ähneln.
DNA Analyse
Eine DNA-Analyse, die zwei krankheitsbedingte Mutationen im MVK-Gen zeigt, wird verwendet, um die Diagnose von HIDS zu bestätigen. In den meisten Fällen hat der Patient zwei verschiedene Mutationen, die als Verbindung bezeichnet werden Heterozygotie.
HIDS-Behandlung
Viele Behandlungen wurden in HIDS ausprobiert, keine mit beständigem Erfolg:
- Colchicin: im Allgemeinen nicht hilfreich, obwohl es Fallberichte über seine erfolgreiche Anwendung gibt.
- Nicht steroidal Antiphlogistikum Die MedikamenteNSAIDs)
- Statine wie Simvastatin, hemmen HMGCoA-Reduktase, die die Produktion von Mevalonsäure reduziert
- Systemisch Kortikosteroide - Eine Einzeldosis zu Beginn eines Anfalls kann die Schwere und Dauer verringern (1 mg / kg).
- Dapson
- Cyclosporin
- Thalidomid
- Intravenös Immunoglobulin (IVIG)
- Biologische Wirkstoffe, einschließlich Anakinra (Interleukin-1 Empfänger Antagonist) und Etanercept (Tumor Nekrose Alpha-Faktor Inhibitor) wurde berichtet, um die Häufigkeit und / oder Schwere von Angriffen in einem 80% zu verringern. Es gab jedoch auch Fälle, in denen diese Mittel häufiger auftraten und / oder Anfälle verlängerten.
Mevalonazidurie
Mevalonazidurie (MIM 610377) beinhaltet das gleiche Gen und Enzym wie HIDS, jedoch ist der resultierende Enzymmangel praktisch vollständig. Der Zustand wird auch als Mevalonatkinase-Mangel bezeichnet. Die bisher identifizierten genetischen Mutationen waren gelegen zu einem Ende des Enzyms. Mevalonazidurie führt zu neurologisch Effekte, die hauptsächlich durch unzureichendes Cholesterin entstehen, das für das Gehirn und das Gehirn notwendig ist Nerv Entwicklung.
Patienten mit Mevalonazidurie erleben die gleichen fieberhaften Episoden wie bei HIDS, entwickeln sich aber auch tiefgreifend Entwicklung Verzögerung, Netzhaut Dystrophie (Sehstörungen) und Wasserfälle, weiche Gesichtsbehandlung Missbildungenund vergrößerte Leber / Milz. Die am wenigsten Betroffenen haben geistige Behinderung, Wachstumsverzögerung, progressiv Kleinhirnataxie (Instabilität) und Anämie. In der Kindheit und Jugend entwickeln sich Augenprobleme, einschließlich Katarakte und Uveitis. Myopathie (Muskelschwäche) kann auftreten. Bei stark Betroffenen ist die Mevalonazidurie im Säuglingsalter normalerweise tödlich.
Zu allen Zeiten werden im Urin hohe Mevalonsäurespiegel festgestellt.
Genetisch Familien mit einem betroffenen Kind sollten beraten werden und vorgeburtlich Beweise sollten berücksichtigt werden.