Hiperinmunoglobulinemia D con Síndrome de Fiebre Periódica (HIDS): Una Visión Integral
Definiendo el Síndrome de Hiper-IgD (HIDS)
La hiperinmunoglobulinemia D con síndrome de fiebre periódica (MIM 260920), conocida popularmente como síndrome de hiper-IgD o HIDS, es un trastorno autoinflamatorio hereditario clasificado como raro. Este síndrome se manifiesta a través de episodios recurrentes que incluyen fiebre, erupciones cutáneas, dolor abdominal significativo, cefaleas y la aparición de ganglios linfáticos agrandados, manifestándose típicamente desde la infancia.
Una forma más grave y potencialmente mortal de esta afección es la aciduria mevalónica.
Epidemiología y Factores de Riesgo del HIDS
El HIDS se observa predominantemente concentrado en dos regiones del norte de Europa: los Países Bajos, donde se localiza más del 50% de los casos notificados, y el norte de Francia. No obstante, se han documentado cerca de 200 casos distribuidos en diversas etnias a nivel mundial.
Generalmente, la sintomatología del HIDS emerge antes de cumplir el primer año de vida, con una edad mediana de aparición fijada en los 6 meses. Sin embargo, establecer un diagnóstico preciso es un proceso complejo que con frecuencia se prolonga durante varios años.
El HIDS se hereda bajo un patrón autosómico recesivo, lo que implica que el individuo debe heredar dos copias del gen defectuoso, una procedente de cada progenitor, quienes generalmente son portadores no afectados de la condición.
Biología Molecular y Fundamentos Genéticos
El gen MVK, asociado al HIDS, se localiza en el cromosoma 12 (12q24) y es el responsable de codificar la enzima mevalonato quinasa. Se ha identificado que mutaciones en este gen están presentes en aproximadamente el 75% de los casos típicos. La mutación V377I es la variante más comúnmente reportada.
En los pacientes con HIDS, las alteraciones genéticas provocan una actividad enzimática reducida, situándose entre el 5% y el 10% de los niveles normales. La aciduria mevalónica representa la presentación más severa, donde la actividad de la enzima es prácticamente indetectable (como se detallará más adelante).
La enzima mevalonato quinasa desempeña un rol crucial en la vía isoprenoide, esencial para la biosíntesis del colesterol. La deficiencia enzimática resultante conduce a la acumulación significativa de ácido mevalónico y a un incremento peligroso en los niveles de interleucina 1.
Manifestaciones Clínicas Detalladas del HIDS
Los episodios agudos y febriles característicos del Síndrome de Hiper-IgD comienzan a manifestarse durante la niñez. Las principales características clínicas observadas incluyen:
- Fiebre elevada.
- Presencia de una erupción maculopapular no migratoria.
- Dolores de cabeza intensos.
- Linfadenopatía dolorosa (agrandamiento de ganglios linfáticos) en la región cervical.
- Dolor abdominal y articular significativo.
La duración promedio de estos ataques se extiende hasta una semana, siendo periodos más largos que los observados en la Fiebre Mediterránea FamiliarQue es familiar Mediterráneo fiebre?La fiebre mediterránea familiar es hereditaria autoinflamatorio síndrome caracterizado por recurrente episodios cortos de fiebre alta asociados con dolor abdominal, inflamación de articulaciones y otras partes del cuerpo y piel erupción. Si no se trata, amilosis comúnmente se desarrolla y puede tener un desenlace fatal. La fiebre mediterránea familiar es el más común de los síndromes de fiebre periódica.En la fiebre mediterránea familiar tipo 2, la más. Estos brotes tienden a repetirse cada uno o dos meses.
A continuación, se detallan las características específicas de los episodios agudos y recurrentes del HIDS en la siguiente tabla descriptiva.
| Síntoma | Características |
|---|---|
| Fiebre |
|
Para ofrecer una comprensión completa del HIDS, es fundamental investigar en profundidad estas manifestaciones clínicas y su manejo diagnóstico.
| Manifestación Clínica | Detalles Relevantes |
|---|---|
| Fiebre | Febrícula persistente con picos que duran varios días; precedentes y malestar generalizados. |
| Erupción cutánea | Afecta hasta el 80% de los casos; presenta varias formas clínicas (descritas más adelante). |
| Cefalea | De naturaleza inespecífica. |
| Linfadenopatía Cervical | Presentación característica; es bilateral, frecuentemente dolorosa. |
| Dolor Abdominal | Puede ser grave, asociado a diarrea y vómitos; en casos raros, puede progresar a peritonitis. |
| Artralgias y Artritis | Artralgia (dolor) o Artritis (inflamación); más prevalente en pacientes jóvenes. Presentación simétrica, poliarticular y no destructiva. Los síntomas articulares generalmente coinciden con el dolor abdominal y se resuelven lentamente después. |
| Hepatomegalia y Esplenomegalia | Agrandamiento del hígado y el bazo (hepatoesplenomegalia); presente en aproximadamente el 50% de los niños afectados. |
| Tendinitis |
Signos Dermatológicos en el HIDS
La afectación cutánea es común, presentándose en hasta el 80% de los pacientes con HIDS. Se han documentado diversas formas de erupciones o erupciones en este síndrome, las cuales tienden a desaparecer gradualmente una vez que el episodio febril ha remitido.
Las manifestaciones cutáneas más frecuentes en el HIDS incluyen:
- Lesiones planas diminutas (máculas).
- Bultos elevados (pequeñas pápulas o nódulos de mayor tamaño).
- Erupción con características similares al sarampión
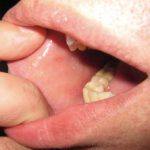 Sarampión: Causas, Síntomas y Riesgos de una Infección Altamente Contagiosa Si sospecha de un diagnóstico confirmado de sarampión, puede enviar sus fotografías relevantes a DermNet para su revisión. El sarampión, también conocido por otros nombres como sarampión inglés, rubéola o morbilli, es provocado por un virus sumamente infección— que se manifiesta con fiebre alta y una característica erupción cutánea. Es fundamental destacar que el sarampión es catalogado como una enfermedad más (morbiliforme).
Sarampión: Causas, Síntomas y Riesgos de una Infección Altamente Contagiosa Si sospecha de un diagnóstico confirmado de sarampión, puede enviar sus fotografías relevantes a DermNet para su revisión. El sarampión, también conocido por otros nombres como sarampión inglés, rubéola o morbilli, es provocado por un virus sumamente infección— que se manifiesta con fiebre alta y una característica erupción cutánea. Es fundamental destacar que el sarampión es catalogado como una enfermedad más (morbiliforme). - Erupción con apariencia de ronchas (urticaria).
Entre las presentaciones cutáneas menos frecuentes o raras, se encuentran:
- Púrpura de Henoch-Schönlein
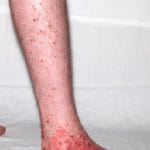 Comprendiendo la Púrpura de Henoch-Schönlein (PHS) La Púrpura de Henoch-Schönlein (PHS) es una manifestación de vasculitis leucocitoclástica o inflamación de vasos sanguíneos pequeños, la cual se diagnostica con mayor frecuencia en la población pediátrica, aunque no es exclusiva de ella. En ocasiones, también es referida como púrpura anafilactoide. La PHS surge de la inflamación que afecta los pequeños vasos sanguíneos localizados en la piel y otros tejidos del cuerpo. Aunque más (púrpura
Comprendiendo la Púrpura de Henoch-Schönlein (PHS) La Púrpura de Henoch-Schönlein (PHS) es una manifestación de vasculitis leucocitoclástica o inflamación de vasos sanguíneos pequeños, la cual se diagnostica con mayor frecuencia en la población pediátrica, aunque no es exclusiva de ella. En ocasiones, también es referida como púrpura anafilactoide. La PHS surge de la inflamación que afecta los pequeños vasos sanguíneos localizados en la piel y otros tejidos del cuerpo. Aunque más (púrpura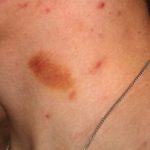 Comprendiendo la Púrpura: Definición, Tipos y Manifestaciones Clínicas La púrpura se define médicamente como la alteración de la coloración de la piel o las membranas mucosas, originada por la extravasación de sangre desde pequeños vasos sanguíneos. Esta aparición cutánea funge como una señal de advertencia indicativa de una posible disfunción en la hemostasia (coagulación) o en la integridad estructural de los capilares y vasos. • Las petequias se caracterizan por más).
Comprendiendo la Púrpura: Definición, Tipos y Manifestaciones Clínicas La púrpura se define médicamente como la alteración de la coloración de la piel o las membranas mucosas, originada por la extravasación de sangre desde pequeños vasos sanguíneos. Esta aparición cutánea funge como una señal de advertencia indicativa de una posible disfunción en la hemostasia (coagulación) o en la integridad estructural de los capilares y vasos. • Las petequias se caracterizan por más). - Eritema elevatum diutinum.
- Petequias (pequeños puntos hemorrágicos o de color púrpura).
- Eritema nodoso.
Además, hasta el 50% de los pacientes experimenta úlceras aftosas orales y/o vaginales.
Factores Desencadenantes de los Ataques
Los episodios sintomáticos agudos del HIDS pueden ser provocados por varios elementos identificables, entre ellos:
- Vacunaciones: Más del 50% de los individuos reportan al menos un episodio durante la infancia posterior a recibir una vacuna.
- Infecciones.
- Estrés significativo, tanto físico como emocional.
- Traumatismos, incluyendo intervenciones quirúrgicas.
¿Cuál es el Pronóstico para los Pacientes con HIDS?
Generalmente, existe una evolución favorable con la edad; los ataques tienden a ser menos frecuentes y su severidad disminuye en la edad adulta. Es fundamental destacar que entre los episodios agudos, la salud general del paciente se mantiene normal.
Sin embargo, un reducido subgrupo de individuos que padecen esta afección puede desarrollar anormalidades neurológicas en la edad adulta, manifestándose de manera similar a la aciduria mevalónica (condición discutida en otros contextos).
A diferencia de la fiebre mediterránea familiar, la presentación de amiloidosis es excepcional en el HIDS, afectando a menos del 3% de los casos. La esperanza de vida suele ser normal para la mayoría de los pacientes, aunque...
Esto puede verse afectado por fallo renal debido a amiloidosis o infecciones graves.
Diagnóstico del HIDS (Deficiencia de Mevalonato Quinasa)
Los ataques febriles agudos recurrentes, especialmente cuando carecen de una causa infecciosa o autoinmune clara, exigen una evaluación diagnóstica exhaustiva.
Criterios Clínicos Esenciales
Además de documentar los episodios febriles recurrentes característicos, los criterios diagnósticos clínicos fundamentales incluyen:
- Inicio de los síntomas antes de los 5 años de edad.
- Duración de los episodios febriles limitada a menos de 14 días.
La presencia de estas características es crucial, ya que las mutaciones en el gen MVK resultan menos probables en su ausencia.
Evaluación de los Niveles de Inmunoglobulina D (IgD)
Es común observar niveles elevados de IgD en numerosos pacientes, aunque este hallazgo no es universal, particularmente en niños menores de 3 años. Estos niveles tienden a elevarse tanto durante un episodio febril como entre estos. Es fundamental interpretar esta elevación con cautela, ya que los niveles de IgD también pueden aumentar en otros síndromes febriles periódicos, como la fiebre mediterránea familiar y el TRAPS, así como en otras afecciones crónicas inflamatorias.
Pruebas de Laboratorio Útiles Adicionales
Durante un ataque agudo, el análisis de ácidos orgánicos en orina frecuentemente revela niveles elevados de ácido mevalónico.
Además, durante el curso de un episodio febril, se observan aumentos significativos en el recuento de glóbulos blancos (leucocitosis), la velocidad de sedimentación globular (VSG o ESR), la proteína C-reactiva (CRP) y la amiloide sérica A (SAA). Los niveles séricos de IgA también pueden experimentar un incremento.
Las pruebas que emplean ensayos radiométricos pueden confirmar una actividad reducida de la mevalonato quinasa, ya sea en glóbulos blancos o en cultivos de fibroblastos.
Una biopsia cutánea podría revelar una leve vasculitis con potencial de extensión profunda. Los cambios morfológicos pueden asemejarse a los observados en la enfermedad de Sweet o la celulitis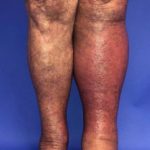 ¿Qué es la celulitis?La celulitis es una bacteriano infección de la baja dermis y subcutáneo tejido. Resulta en un localizado área de piel roja, dolorosa e hinchada, y sistémico síntomas. Se experimentan síntomas similares con la infección más superficial, la erisipela, por lo que la celulitis y la erisipela a menudo se consideran juntas. Celulitis Celulitis de la pierna izquierda ¿Quién tiene celulitis?La celulitis afecta a personas de todas las más.
¿Qué es la celulitis?La celulitis es una bacteriano infección de la baja dermis y subcutáneo tejido. Resulta en un localizado área de piel roja, dolorosa e hinchada, y sistémico síntomas. Se experimentan síntomas similares con la infección más superficial, la erisipela, por lo que la celulitis y la erisipela a menudo se consideran juntas. Celulitis Celulitis de la pierna izquierda ¿Quién tiene celulitis?La celulitis afecta a personas de todas las más.
Confirmación Mediante Análisis de ADN
El diagnóstico definitivo de HIDS se confirma mediante el análisis de ADN que demuestra la presencia de dos mutaciones patogénicas asociadas a la enfermedad en el gen MVK. En la mayoría de los casos, el individuo presenta dos alelos mutados diferentes, una condición conocida como heterocigosidad compuesta.
Opciones de Tratamiento para el HIDS
Se ha investigado una amplia gama de tratamientos para el HIDS, pero ninguno ha demostrado una eficacia universal o consistente:
- ColchicinaPropiedades y Uso de la Colchicina en Dermatología La colchicina es un fármaco ancestral, derivado del azafrán de otoño, o *Colchicum autumnale* (una planta venenosa originaria de Europa). Históricamente, su aplicación principal ha sido el manejo de la gota. Aunque no está formalmente indicada ni aprobada para el tratamiento de afecciones dermatológicas, la colchicina se ha prescrito con éxito para ciertas patologías cutáneas debido a sus notables propiedades inmunológicas y más: Generalmente ineficaz, aunque se han reportado casos aislados de éxito.
- Fármacos antiinflamatorios no esteroideos (AINEs).
- Estatinas (ej., simvastatina): Actúan al inhibir la HMGCoA-reductasa, disminuyendo la síntesis de ácido mevalónico.
- Corticosteroides sistémicos: Una dosis única al inicio de un ataque puede mitigar su severidad y duración (administración recomendada de 1 mg/kg).
- Dapsona```html Información Esencial sobre la Dapsona: Usos, Monitoreo y Efectos Secundarios La dapsona, un antibiótico perteneciente a la clase de las sulfonas, ha sido un tratamiento establecido y disponible durante muchos años, especialmente para la lepra. En el contexto de Nueva Zelanda, este medicamento se presenta comúnmente en tabletas de 25 mg y 100 mg. Usos Dermatológicos de la Dapsona La dapsona es fundamental en el manejo de diversas afecciones más.
- Ciclosporina
 Comprendiendo la Ciclosporina: Usos e Indicaciones Terapéuticas La ciclosporina es un fármaco inmunosupresor fundamental, empleado principalmente en el tratamiento de diversas enfermedades inflamatorias graves. Su aplicación se extiende a condiciones que afectan tanto la piel como otros órganos internos del cuerpo. En Nueva Zelanda, este tratamiento está accesible y completamente financiado en sus formulaciones de tabletas y líquido oral. Adicionalmente, existen versiones oftálmicas (gotas para los ojos) e inyectables, aunque más.
Comprendiendo la Ciclosporina: Usos e Indicaciones Terapéuticas La ciclosporina es un fármaco inmunosupresor fundamental, empleado principalmente en el tratamiento de diversas enfermedades inflamatorias graves. Su aplicación se extiende a condiciones que afectan tanto la piel como otros órganos internos del cuerpo. En Nueva Zelanda, este tratamiento está accesible y completamente financiado en sus formulaciones de tabletas y líquido oral. Adicionalmente, existen versiones oftálmicas (gotas para los ojos) e inyectables, aunque más. - TalidomidaTalidomida: Descifrando sus Usos Terapéuticos Actuales y Mecanismo de Acción A finales de la década de 1950, la talidomida irrumpió en el mercado como un medicamento revolucionario, prometiendo alivio contra el insomnio y las náuseas, especialmente en mujeres embarazadas. Trágicamente, su promesa se desvaneció rápidamente cuando se documentaron numerosos y graves defectos congénitos en los neonatos expuestos al fármaco in utero. Este descubrimiento llevó a su restricción mundial inmediata. Sin más.
- Inmunoglobulina intravenosa (IVIG).
- Agentes biológicos: Incluyen anakinraAnakinra: Agente Biológico para Enfermedades Reumatoides y Dermatológicas Anakinra es un agente biológico recombinante empleado primordialmente en el tratamiento de enfermedades reumáticas, como la artritis reumatoide. Recientemente, su utilidad se ha expandido, demostrando ser eficaz en el manejo de diversas afecciones cutáneas inflamatorias. Composición y Desarrollo de Anakinra Anakinra funciona como un antagonista del receptor de interleucina-1 (IL-1Ra), generado mediante tecnología de ADN recombinante, utilizando un sistema de expresión bacteriano más (un antagonista del receptor de interleucina-1) y etanercept (un inhibidor del factor alfa de necrosis tumoral). Se ha documentado que estos reducen la frecuencia y/o gravedad de los ataques hasta en un 80%. No obstante, también existen informes donde estos tratamientos han provocado un aumento o una prolongación de los episodios.
Aciduria Mevalónica (MIM 610377)
La aciduria mevalónica comparte el gen afectado y la misma enzima que el HIDS; sin embargo, la deficiencia enzimática resultante es casi total o completa. Esta condición también es denominada deficiencia de mevalonato quinasa. Las mutaciones genéticas identificadas hasta la fecha suelen estar localizadas en un extremo de la estructura de la enzima. La aciduria mevalónica produce efectos neurológicos significativos, derivados principalmente de la producción inadecuada de colesterol, un componente esencial para el cerebro y el desarrollo de los nervios.
Los pacientes con aciduria mevalónica experimentan los mismos episodios febriles que en el HIDS, pero adicionalmente desarrollan un profundo retraso en el desarrollo, distrofia retiniana (con defectos visuales asociados) y cataratas. Presentan también deformidades faciales leves y hepatomegalia/esplenomegalia (aumento del tamaño del hígado y el bazo). Los individuos menos severamente afectados pueden manifestar retraso mental leve, detención del crecimiento, ataxia cerebelosa progresiva (inestabilidad al caminar) y anemia. Durante la infancia y adolescencia, surgen complicaciones oculares, incluyendo cataratas y uveítis. Asimismo, puede desarrollarse miopatía (debilidad muscular). En los casos graves, la aciduria mevalónica resulta fatal durante la primera década de vida.
Los niveles elevados de ácido mevalónico se detectan consistentemente en muestras de orina en todo momento.
A las familias con un niño afectado se les debe ofrecer asesoramiento genético, y debe considerarse la realización de pruebas prenatales.


