Tildrakizumab (Ilumya™): Revisión Técnica y Datos de Eficacia Clínica
En marzo de 2018, tildrakizumabTildrakizumab (Ilumya™): Mecanismo de Acción y Pautas de Administración para la Psoriasis en Placas En marzo de 2018, el tratamiento biológico tildrakizumab, comercializado bajo el nombre de Ilumya™ por Sun Pharma India, recibió la aprobación de la Administración de Drogas y Alimentos de EE. UU. (FDA). Esta autorización se centró en el manejo de la psoriasis en placas de intensidad moderada a grave en pacientes que son candidatos a terapias más (Ilumya™; Sun Pharma, Mumbai, India) obtuvo la aprobación de la Administración de Drogas y Alimentos de EE. UU. (FDA) para el tratamiento de la psoriasis en placas de moderada a grave en pacientes candidatos a terapia sistémica o fototerapiaLa fototerapia constituye el uso estratégico de determinados tipos de radiación electromagnética con el fin de tratar diversas afecciones dermatológicas. Esta técnica emplea longitudes de onda específicas para modular la respuesta biológica de la piel. Visite las páginas de DermNet NZ sobre fototerapia para obtener información exhaustiva. Los métodos y aplicaciones principales de la fototerapia incluyen: • Tratamiento del acné mediado por luz azul. • Terapia con radiación ultravioleta B más. Este anticuerpo monoclonal se une selectivamente a la subunidad p19 de la interleucina (IL) 23 e inhibe su interacción con el receptor de IL-23. Sun Pharma licenció este compuesto de Merck and Co. (Nueva Jersey, EE. UU.) en 2014.
Tildrakizumab representa la segunda terapia biológica autorizada que bloquea específicamente la IL-23, una citocina fundamental en la patogénesis de la psoriasis en placas.
La aprobación de la FDA para tildrakizumab se fundamentó en los sólidos datos de seguridad y eficacia recopilados durante los ensayos clínicos de fase III reSURFACE. Estos estudios compararon tildrakizumab contra placebo y etanercept. En ambos ensayos, tildrakizumab demostró mejoras estadísticamente significativas frente a los grupos control en la semana 12, evaluadas mediante el Índice de Sensibilidad del Área de Psoriasis (PASI) y las puntuaciones de Evaluación Global del Médico (PGA).
En Europa, Almirall posee los derechos para el desarrollo y comercialización de este fármaco, habiendo presentado la solicitud de aprobación regulatoria ante la Agencia Europea de Medicamentos en marzo de 2017.
Es importante notar que, actualmente, tildrakizumab no se encuentra disponible para su uso en Nueva Zelanda.
Análisis de la Evidencia de Ensayos Clínicos para Tildrakizumab
La decisión regulatoria de la FDA se basó en los resultados obtenidos de dos ensayos clínicos cruciales de fase III:
- reSURFACE 1: Evaluó la eficacia y el perfil de seguridad de tildrakizumab en comparación directa con un placebo.
- reSURFACE 2: Comparó la eficacia y seguridad de tildrakizumab contra etanercept y placebo.
Resultados Detallados del Ensayo reSURFACE 1
- El estudio reSURFACE 1 se configuró como un ensayo multicéntrico, doble ciego, aleatorizado y de grupos paralelos.
- Reclutó a 1.772 pacientes que padecían psoriasis de moderada a grave. Los participantes fueron asignados aleatoriamente a recibir tildrakizumab 100 mg (n = 309), tildrakizumab 200 mg (n = 308) o placebo (n = 155) en las semanas 0, 4 y 16.
- Los pacientes que inicialmente recibieron placebo fueron cruzados al régimen de tildrakizumab (100 mg o 200 mg) en las semanas 12 y 16.
- Los criterios de valoración primarios definieron el éxito como el porcentaje de pacientes en cada grupo que alcanzaron una mejora del 75% en la puntuación PASI (PASI 75) y una puntuación PGA (Evaluación global de médicos) de 0 o 1 junto con una reducción de más de 2 puntos desde el valor basal a las 12 semanas.
- A la semana 12, un porcentaje significativamente superior de pacientes en ambos grupos de tildrakizumab logró los criterios PASI 75 y PGA (0 o 1) en comparación con el grupo placebo (PASI 75: 100 mg: 64%, 200 mg: 62%, placebo: 6%; p < 0,001; PGA puntuación de 0 o 1: 100 mg: 59%, 200 mg: 58%, placebo: 7%; p <0,001).
Resultados del Ensayo reSURFACE 2
- El estudio reSURFACE 2 incluyó a 1.090 pacientes con la misma indicación de psoriasis moderada a grave. Estos fueron asignados aleatoriamente a recibir placebo, tildrakizumab 100 mg o 200 mg (administrados en las semanas 0, 4 y 16), o etanercept 50 mg dos veces por semana durante 12 semanas, seguido de una dosis semanal hasta la semana 28.
- Los puntos finales coprimarios establecidos fueron análogos a los del estudio reSURFACE 1.
- En la semana 12, una proporción significativamente mayor de pacientes en los grupos tratados con tildrakizumab alcanzó PASI 75 en comparación con los grupos de placebo y etanercept (100 mg: 61%, 200 mg: 66%, etanercept: 48%, placebo: 6%; p < 0,0001 para ambas comparaciones contra placebo; p < 0,0001 para 200 mg versus etanercept y p = 0,0010 para 100 mg versus etanercept).
- Este patrón de superioridad se mantuvo para la proporción de pacientes que alcanzaron una puntuación PGA de 0 o 1 en la semana 12 (tildrakizumab 100 mg: 55%, tildrakizumab 200 mg: 59%, placebo: 4%; p < 0,001) y se extendió hasta la semana 28 (tildrakizumab 100 mg: 66%, tildrakizumab 200 mg: 71%, etanercept: 48%; p < 0,001).
- El porcentaje de pacientes que experimentaron al menos un acontecimiento adverso en cada grupo de tratamiento hasta la semana 12 fue el siguiente: tildrakizumab
En resumen, los datos robustos de reSURFACE 1 y 2 confirman la alta eficacia de tildrakizumab en la inducción y mantenimiento de la mejoría en pacientes con psoriasis en placas moderada a grave, superando consistentemente las respuestas observadas con placebo y, en ciertos parámetros, con etanercept en las primeras 12 semanas. Estos ensayos son el pilar de la reciente aprobación regulatoria en EE. UU.
- 100 mg: 44,3%, tildrakizumab 200 mg: 49,4%, placebo: 55,1%, etanercept: 54,0%. Se registró un fallecimiento en un paciente del grupo de tildrakizumab 100 mg que padecía cardiomiopatía alcohólica y esteatosis hepática; la causa de la muerte fue clasificada como indeterminada.
Evaluación de Reacciones Adversas en Ensayos Clínicos
Dado que los ensayos clínicos se conducen bajo condiciones sumamente variables, las tasas de reacción adversa observadas en estos estudios farmacológicos no deben compararse directamente con las tasas obtenidas en los ensayos de otros medicamentos, y es posible que no reflejen con exactitud las frecuencias vistas en la práctica clínica rutinaria.
Para determinar la seguridad del tildrakizumab, se combinaron los datos obtenidos de los ensayos de fase III, aleatorizados y controlados con placebo. La siguiente tabla resume las reacciones adversas que se manifestaron en al menos el 1% de los participantes y con una frecuencia superior en el grupo tratado con tildrakizumab 100 mg frente al grupo placebo.
Tabla: Reacciones Adversas Ocurridas en ≥ 1% de Sujetos Tratados con Tildrakizumab
| Reacción Adversa | Tildrakizumab 100 mg (n = 705) | Placebo |
|---|---|---|
| Infecciones del tracto respiratorio superior | 98 (14%) | 41 (12%) |
| Reacciones en el sitio de inyección | 24 (3%) | 7 (2%) |
| Diarrea | 13 (2%) | 5 (1%) |
Las infecciones respiratorias identificadas como reacciones adversas en pacientes que recibieron tildrakizumab incluyeron:
- Nasofaringitis
- Infección del tracto respiratorio superior
- Infección viral del tracto respiratorio superior
- Faringitis.
Las reacciones ocurridas en el sitio de inyección reportadas como efectos adversos en pacientes tratados con tildrakizumab comprendieron:
- Urticaria
- Prurito
- Eritema
- Inflamación
- Edema
- Hinchazón
- Hematomas
- Hematoma
- Hemorragia.
Las reacciones adversas que se manifestaron en menos del 1% del grupo de tildrakizumab, pero con una tasa superior a la del grupo placebo, fueron:
- Mareos
- Dolor en las extremidades.
En sujetos tratados con tildrakizumab se han documentado casos de angioedema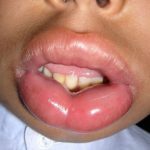 Comprendiendo el Angioedema: Definición y Diferencias Clave con la Urticaria El angioedema representa una reacción cutánea significativa, a menudo comparable en apariencia a la urticaria. Su característica principal es una hinchazón repentina y típicamente breve que afecta tanto la piel como las membranas mucosas. Si bien cualquier parte del cuerpo puede verse comprometida, la localización más habitual para esta inflamación es el contorno de los ojos y los labios. En más y urticaria aguda
Comprendiendo el Angioedema: Definición y Diferencias Clave con la Urticaria El angioedema representa una reacción cutánea significativa, a menudo comparable en apariencia a la urticaria. Su característica principal es una hinchazón repentina y típicamente breve que afecta tanto la piel como las membranas mucosas. Si bien cualquier parte del cuerpo puede verse comprometida, la localización más habitual para esta inflamación es el contorno de los ojos y los labios. En más y urticaria aguda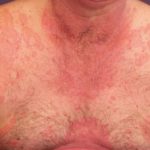 ```html Entendiendo la Urticaria: Síntomas, Tipos y Causas La urticaria se manifiesta a través de la aparición de habones (ronchas), o bien mediante angioedema (hinchazón más profunda, presente en el 10% de los casos), o en ocasiones ambas afecciones simultáneamente (40% de los casos). El término médico proviene de la ortiga común europea, *Urtica dioica*. Una roncha, también conocida como habón, es una elevación superficial de la piel, del mismo más durante los ensayos clínicos. Si se presenta una reacción grave de hipersensibilidad, es imperativo suspender el medicamento de inmediato e instaurar la terapia apropiada.
```html Entendiendo la Urticaria: Síntomas, Tipos y Causas La urticaria se manifiesta a través de la aparición de habones (ronchas), o bien mediante angioedema (hinchazón más profunda, presente en el 10% de los casos), o en ocasiones ambas afecciones simultáneamente (40% de los casos). El término médico proviene de la ortiga común europea, *Urtica dioica*. Una roncha, también conocida como habón, es una elevación superficial de la piel, del mismo más durante los ensayos clínicos. Si se presenta una reacción grave de hipersensibilidad, es imperativo suspender el medicamento de inmediato e instaurar la terapia apropiada.
Las tasas de infecciones graves fueron de ≤ 0,3% tanto para el grupo de tildrakizumab como para el grupo placebo. Por lo tanto, el tratamiento con tildrakizumab no debe iniciarse en pacientes con infecciones activas clínicamente significativas hasta que dichas infecciones se resuelvan o reciban un tratamiento adecuado.
Tildrakizumab: Perspectivas y Futuro
Los ensayos clínicos de fase III con tildrakizumab han demostrado resultados muy alentadores respecto a su eficacia y perfil de seguridad a corto plazo. No obstante, es fundamental contar con evidencia adicional y estudios más amplios y prolongados para confirmar estos hallazgos.
Se requieren estudios comparativos con ensayos controlados aleatorios frente a tratamientos establecidos como ustekinumab y guselkumab Guselkumab: Tratamiento Biológico Avanzado para la Psoriasis Guselkumab es un tratamiento biológico avanzado diseñado específicamente para el manejo de la psoriasis en placas de moderada a grave. La Administración de Alimentos y Medicamentos de EE. UU. (FDA) ha otorgado aprobación a guselkumab (comercializado como Tremfya™ por Janssen Biotech, PA, EE. UU.) para tratar a adultos que sufren de **psoriasis en placas** moderada a grave, siempre que califiquen para recibir terapia más para evaluar la eficacia relativa de tildrakizumab. Una ventaja destacada del tildrakizumab reside en su régimen de mantenimiento cada 12 semanas, similar al de ustekinumab, lo cual previsiblemente podría mejorar la adherencia del paciente al tratamiento.
Guselkumab: Tratamiento Biológico Avanzado para la Psoriasis Guselkumab es un tratamiento biológico avanzado diseñado específicamente para el manejo de la psoriasis en placas de moderada a grave. La Administración de Alimentos y Medicamentos de EE. UU. (FDA) ha otorgado aprobación a guselkumab (comercializado como Tremfya™ por Janssen Biotech, PA, EE. UU.) para tratar a adultos que sufren de **psoriasis en placas** moderada a grave, siempre que califiquen para recibir terapia más para evaluar la eficacia relativa de tildrakizumab. Una ventaja destacada del tildrakizumab reside en su régimen de mantenimiento cada 12 semanas, similar al de ustekinumab, lo cual previsiblemente podría mejorar la adherencia del paciente al tratamiento.
A medida que avanza la comprensión de la inmunopatogenia de la psoriasis, el foco se ha desplazado hacia blancos terapéuticos más específicos. Actualmente, se considera que el eje IL-23 / IL-17 juega un papel fundamental en la patogénesis de la psoriasis.
Los medicamentos que actúan sobre la subunidad p40, común a IL-12 e IL-23 (como ustekinumab), han demostrado una potente actividad clínica. No obstante, la selectividad por IL-23p19 podría ofrecer beneficios superiores tanto en eficacia como en perfil de seguridad en comparación con el bloqueo anti-p40. Al intervenir antes en la cascada de citoquinas IL-23 / IL-17, es probable que se consiga una reducción en la expresión de múltiples citoquinas proinflamatorias en los queratinocitos, incluyendo IL-17F, IL-21, IL-22, además de IL-17A.
Basándose en este conocimiento, la focalización selectiva de la subunidad IL-23p19 se ha consolidado como una opción terapéutica atractiva, impulsando el desarrollo de una nueva clase de agentes biológicos (ej. guselkumab y tildrakizumab) para manejar la psoriasis de moderada a grave. Hasta la fecha, los datos de seguridad sugieren que estos fármacos podrían estar asociados con una menor incidencia de algunos efectos adversos observados con el bloqueo de IL-17A (fármacos como secukinumab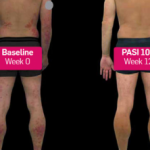 Secukinumab (Cosentyx®): Mecanismo de Acción y Eficacia Clínica en Dermatología El secukinumab pertenece a la vanguardia de tratamientos biológicos, específicamente categorizado como un anticuerpo monoclonal. Este medicamento, comercializado bajo la marca Cosentyx® (Novartis AG) [1], representa un avance crucial para el manejo de diversas patologías inmunomediadas y afecciones dermatológicas. Actualmente, el secukinumab está formalmente indicado para el control de la psoriasis en placas de moderada a grave en pacientes adultos más y brodalumab
Secukinumab (Cosentyx®): Mecanismo de Acción y Eficacia Clínica en Dermatología El secukinumab pertenece a la vanguardia de tratamientos biológicos, específicamente categorizado como un anticuerpo monoclonal. Este medicamento, comercializado bajo la marca Cosentyx® (Novartis AG) [1], representa un avance crucial para el manejo de diversas patologías inmunomediadas y afecciones dermatológicas. Actualmente, el secukinumab está formalmente indicado para el control de la psoriasis en placas de moderada a grave en pacientes adultos más y brodalumab Brodalumab (Siliq™): Tratamiento de la Psoriasis en Placas y Consideraciones de Seguridad En febrero de 2017, la Administración de Drogas y Alimentos de EE. UU. (FDA) autorizó el uso del biológico brodalumab (Siliq™; comercializado por Valeant Pharmaceuticals, Nueva Jersey, EE. UU.). Este medicamento está aprobado para tratar la psoriasis en pacientes adultos que no han respondido de manera adecuada a las terapias previamente establecidas. Valeant planeaba introducir brodalumab en el más), tales como infecciones mucocutáneas por cándida o la provocación o exacerbación de enfermedad inflamatoria intestinal.
Brodalumab (Siliq™): Tratamiento de la Psoriasis en Placas y Consideraciones de Seguridad En febrero de 2017, la Administración de Drogas y Alimentos de EE. UU. (FDA) autorizó el uso del biológico brodalumab (Siliq™; comercializado por Valeant Pharmaceuticals, Nueva Jersey, EE. UU.). Este medicamento está aprobado para tratar la psoriasis en pacientes adultos que no han respondido de manera adecuada a las terapias previamente establecidas. Valeant planeaba introducir brodalumab en el más), tales como infecciones mucocutáneas por cándida o la provocación o exacerbación de enfermedad inflamatoria intestinal.
Los medicamentos biológicos dirigidos a estas citoquinas y sus receptores han probado ser eficaces y seguros en ensayos clínicos. Además, han demostrado una eficacia superior a los biológicos previos, evidenciado por la alta proporción de pacientes que alcanzan no solo una mejoría del 75% en la puntuación PASI (PASI 75), sino también mejoras significativas del 90% (PASI 90) y 100% (PASI 100).
Las hojas de datos aprobadas por Nueva Zelanda constituyen la fuente oficial y autorizada de información para medicamentos de prescripción, detallando usos aprobados e información de riesgos. Se recomienda consultar la hoja de datos específica de Nueva Zelanda disponible en el Sitio web de Medsafe.
Si usted no reside en Nueva Zelanda, le instamos a consultar con la agencia reguladora de medicamentos de su país para obtener la información más relevante sobre los fármacos (ejemplo: la Administración Australiana de Productos Terapéuticos y la Administración de Drogas y Alimentos de EE. UU.) o con un formulario nacional o estatal aprobado (ejemplo: el Formulario de Nueva Zelanda y Formulario de Nueva Zelanda para niños, y el Formulario nacional británico y Formulario nacional británico para niños).


