Was ist es Amyloidose?
Amyloidose ist der Begriff für eine Gruppe von Krankheiten, bei denen ein oder mehrere Organe des Körpers verschiedene unlösliche Substanzen ansammeln. Protein (Amyloid) in Mengen zu verursachen Funktionsstörung des Organsystems. Häufig betroffene Organe sind Herz, Nieren, Magen-Darm-Trakt, Nervensystem und Haut. Amyloidose der Haut wird genannt Haut- Amyloidose. In diesem Zustand lagern sich Amyloid- oder Amyloid-ähnliche Proteine in der Dermis.
Arten von Amyloidose
Es gibt drei Hauptarten von Amyloidose.
Primär Amyloidose
- Primäre Amyloidose ist eine Störung von Protein Stoffwechsel Es stammt aus dem Knochenmark und ist gelegentlich mit multiplem Myelom assoziiert. Es wird manchmal auch als Amyloidose vom L-Ketten-Amyloid (AL) -Typ bezeichnet.
- Primär systemisch Amyloidose betrifft Herz, Nieren, Leber, Magen-Darm-Trakt und das Zentralnervensystem. Eine Hautbeteiligung tritt bei ungefähr 30 bis 40% von Patienten auf.
- Gelegen Die primäre Amyloidose betrifft bestimmte Organe wie die Haut (primäre kutane Amyloidose), das Herz, die Augen, die Atemwege und die Blase.
Arten der primären lokalisierten kutanen Amyloidose umfassen:
- Flechtenamyloidose
- Makular Amyloidose
- Knoten Amyloidose
- Amyloidose Haut dyschromica.
Sekundäre systemische Amyloidose
- Eine sekundäre systemische Amyloidose tritt bei vielen als Komplikation auf chronisch entzündlich Krankheiten wie Rheumatoide Arthritis und Osteomyelitis.
- Es ist auch als Amyloid A (AA) -Amyloidose bekannt und weltweit die häufigste Form der systemischen Amyloidose.
- Niere, Leber und Milz sind die am stärksten betroffenen Organe bei der sekundären systemischen Amyloidose. Hautbeteiligung ist selten ein Merkmal der Krankheit.
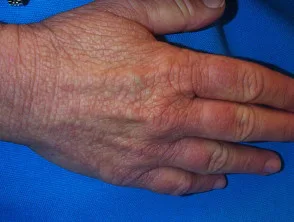
Familie (erblich) Amyloidose
Familiäre Amyloidose ist eine seltene Form der Amyloidose, die am häufigsten vererbt und verursacht wird Kardiomyopathie und Neuropathie im mittleren Alter. Die häufigste Form, bekannt als ATTR-Amyloidose, ist auf a zurückzuführen Mutation in Transthyretin (TTR) Gen im Chromosom 5 und führt zum Versagen hepatisch Transthyretin-Protein.
Was sind die klinischen Merkmale der primären Amyloidose?
Für jede Art der primären Amyloidose werden charakteristische klinische Merkmale gefunden.
Systemische Amyloidose
Die Anzeichen und Symptome einer primären systemischen Amyloidose sind im Allgemeinen nicht spezifisch und umfassen:
- Ermüden
- Gewichtsverlust
- Ödem
- Atembeschwerden
- Benommenheit
- Taubheit, Kribbeln
- Heiserkeit.
Diese können der Diagnose einer Amyloidose um bis zu zwei Jahre vorausgehen. In Verbindung mit den folgenden spezifischen Symptomen sollte jedoch eine systemische Amyloidose als mögliche Diagnose angesehen werden.
- Karpaltunnel Syndrom: Änderung von Median Nerv Funktion im Handgelenk, wenn der Nerv durch den Karpaltunnel geht.
- Haut Verletzungen
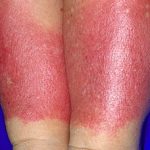
- Die häufigsten Hautbefunde sind lila: Petechien (kleine rote Blutflecken) und Ekchymose (kleine, flache, runde oder unregelmäßige und bläuliche / violette Blutflecken).
- Wachsig Papeln, Knötchen oder Platten kann um die Augenlider, Hals, Leistengegend und gefunden werden anogenital Zone.
- Mit Blut gefüllte Blasen können sich beispielsweise durch Einklemmen der Haut bilden.
- das Nagel kann spröde und spröde seinein Dystrophie).
- Patches Haar Verlust kann entstehendiffus Alopezie).
- Makroglossie: bezieht sich auf eine geschwollene, feste Zunge, die mit Blutflecken, Plaques und Blasen bedeckt ist.
- Ödeme (Entzündungen des Gewebes) entstehen durch Herzinsuffizienz oder nephrotisches Syndrom (Nierenerkrankung).
- Hepatomegalie (vergrößerte Leber).
Systemische Amyloidose
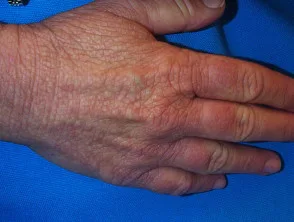
Erbliches Amyloid
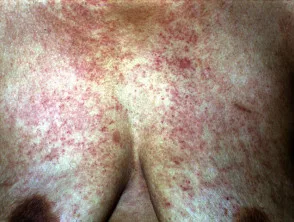
Amyloidosis purpura
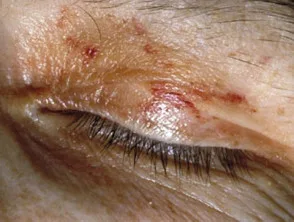
Amyloidosis purpura
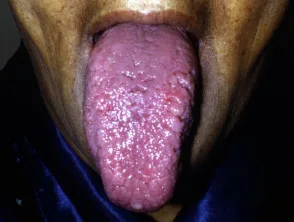
Makroglossie durch systemische Amyloidose
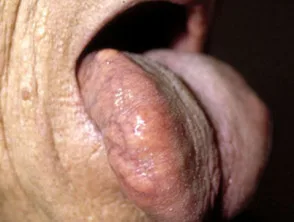
Makroglossie durch systemische Amyloidose
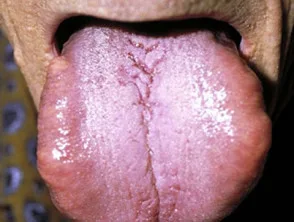
Makroglossie durch systemische Amyloidose
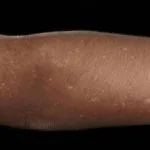
Flechtenamyloidose
- Auch genannt papulös Amyloidose
- Die Flechtenamyloidose ist eine der beiden häufigsten Formen der primären lokalisierten kutanen Amyloidose.
- Es zeigt sich als starker Juckreiz. Eruption an Schienbeinen, Oberschenkeln, Füßen und Unterarmen.
- Läsionen bestehen aus mehreren erhabenen Flecken (Papeln), die sind schuppig und rot / braun.
- Papeln können zusammenkommen in verdickten Platten.
- Es scheint häufiger bei Menschen chinesischer Abstammung und Männern zu sein.
- Es tritt am häufigsten bei Menschen im Alter von 50 bis 60 Jahren auf.
- Kann eine Variante der Flechte sein Simplex.
Flechtenamyloidose
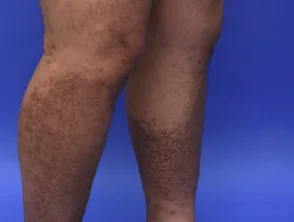
Flechtenamyloidose
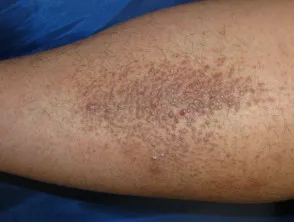
Flechtenamyloidose
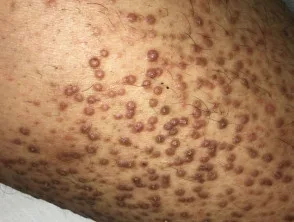
Flechtenamyloidose
Makula-Amyloidose
- Bei der Makula-Amyloidose erscheinen die Läsionen als flache dunkelbraune oder graue Flecken, die zu Flecken dunkler Haut verschmelzen können.
- Der Grad des Juckreizes variiert von leicht bis schwer.
- Die Läsionen sind normalerweise im oberen Rücken zwischen den Schulterblättern, der Brust und manchmal den Armen verteilt.
- Es scheint häufiger bei Asiaten, Südamerikanern und im Nahen Osten zu sein.
- Es tritt normalerweise im frühen Erwachsenenalter auf und betrifft Frauen häufiger als Männer.
- Makula-Amyloidose soll genauso häufig sein wie Flechten-Amyloidose, ist aber aufgrund ihrer vagen Darstellung und Fehldiagnose wahrscheinlich häufiger.
Makula-Amyloidose
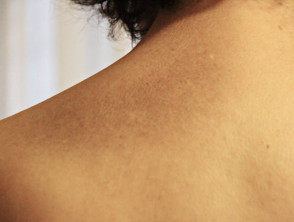
Makula-Amyloidose
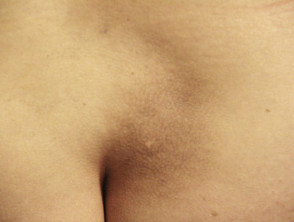
Makula-Amyloidose
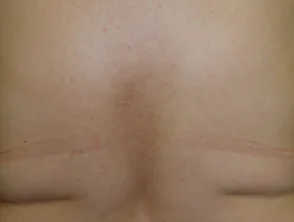
Makula-Amyloidose
Primäre knotenlokalisierte kutane Amyloidose
- Auch bekannt als Amyloidosis cutis nodularis atrophicans und geschwollene Amyloidose
- Die knotige primäre lokalisierte kutane Amyloidose ist die seltenste Form der primären lokalisierten kutanen Amyloidose.
- Einzelne oder mehrere feste Plaques oder Knötchen können am Rumpf, an den Extremitäten, Extremitäten, im Gesicht und an den Genitalien auftreten.
- Die primäre lokalisierte kutane Amyloidose des Knotens ist normalerweise asymptomatisch, und die Patienten suchen wegen ihres Aussehens einen Arzt auf.
- Knötchen können einige Millimeter bis einige Zentimeter groß sein und eine rosa-braune bis rote Farbe haben.
- Die Läsionen neigen dazu, nicht zu ulzerieren, aber einige können reißen oder splittern, insbesondere an den Fußsohlen.
Knotige Amyloidose
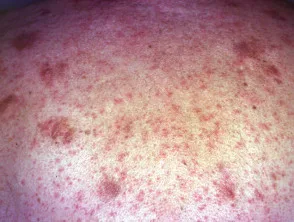
Knotige Amyloidose
Amyloidosis cutis dischromicum
Amyloidose Haut Dyschromie ist eine kürzlich beschriebene Form der primären kutanen Amyloidose, die verursacht Hyperpigmentierung und Hypopigmentierung.
Wie wird die Diagnose einer Amyloidose gestellt?
Die Diagnose der verschiedenen Arten der primären kutanen Amyloidose wird anhand ihres klinischen Erscheinungsbilds und der Merkmale gestellt Histologie beobachtete Veränderungen in der Haut Biopsie. Siehe auch systemische Amyloidose. Pathologie.
Was ist die Behandlung für systemische Amyloidose?
Unspezifische Behandlung
Die Behandlung zielt darauf ab, die Funktion der betroffenen Organe aufrechtzuerhalten. Zum Beispiel kann Nierenversagen mit behandelt werden Dialyse und Herzinsuffizienz mit Diuretika.
Spezifische Behandlung
- Chemotherapie mit Melphalan und Prednison oder Dexamethason (oral Kortikosteroide) können Steuerung systemische Amyloidose.
- Bei sekundärer Amyloidose kann Colchicin hemmen Amyloid Erklärung während die Behandlung gegen die Hauptursache gerichtet ist.
- Gelang es monoklonal Antikörper zur Anwendung bei systemischer Amyloidose gehören Lenalidomid und Bortezomib.
- Knochenmark Abtragung kann unternommen werden, um die zu beseitigen Plasma Zellen und wird von autologen Stammzellen gefolgt Transplantation.
- Die hereditäre ATTR-Amyloidose wird mit Diflunisal oder Tafamidis (die das abnormale Transthyretin-Protein stabilisiert) behandelt. Die Lebertransplantation ist den am stärksten betroffenen Personen vorbehalten.
Prognose
Die meisten Patienten, die keine Behandlung erhalten, sterben ein bis zwei Jahre nach der Diagnose einer primären systemischen Amyloidose aufgrund von Herz- oder Nierenversagen.
Was ist die Behandlung für primäre kutane Amyloidose?
Die Behandlung von Flechten und Makula-Amyloidose konzentriert sich auf die Linderung von Juckreiz. Beruhigende Antihistaminika können mäßig wirksam sein. Aktuell und intraläsionale Steroide können eine gewisse Linderung bringen, wenn sie mit anderen Behandlungen verwendet werden. Andere Behandlungen umfassen topisches Dimethylsulfoxid (DMSO) und Phototherapie (UVB oder PUVA).
Die chirurgische Behandlung zur Entfernung von Amyloidablagerungen umfasst Sein Verdampfung, Dermabrasion und Exzision individuelle Verletzungen.
Das Hauptziel der Behandlung der knotigen primären lokalisierten kutanen Amyloidose ist die Verbesserung des Aussehens. Verschiedene Methoden werden verwendet und umfassen Antibiotika, topische und intraläsionale Kortikosteroide, Kryotherapie, Dermabrasion, Rasieren, Kürettage und Elektrodenkation, Kohlendioxidlaser und gepulster Farbstofflaser.
Primäre kutane Amyloidose neigt dazu bestehen bleiben oder wiederholen nach der Behandlung, bleibt aber auf die Haut beschränkt und entwickelt sich nicht zu einer systemischen Erkrankung.