Die Internationale Klassifikation der Ehlers-Danlos-Syndrome 2017
Siehe Ehlers–Danlos Syndrom.
Die spezifischen Subtypen des Ehlers-Danlos-Syndroms (EDS) oder Ehlers-Danlos-Syndroms werden im Folgenden beschrieben. [1,2]. Sie beinhalten:
- Klassisches Ehlers-Danlos-Syndrom (cEDS)
- Ehlers-Danlos-Syndrom vom klassischen Typ (clEDS)
- Herzklappen-Ehlers-Danlos-Syndrom (cvEDS)
- Gefäß Ehlers-Danlos-Syndrom (vEDS)
- Hypermobiles Ehlers-Danlos-Syndrom (hEDS)
- Arthrochalasie-Ehlers-Danlos-Syndrom (aEDS)
- Dermatosparaxis Ehlers-Danlos-Syndrom (dEDS)
- Kyphoskoliotisches Ehlers-Danlos-Syndrom (kEDS)
- Fragil Hornhaut Syndrom (BCS)
- Ehlers-Danlos-spondylodysplastisches Syndrom (spEDS)
- Ehlers-Danlos muskulokontrakturales Syndrom (mcEDS)
- Myopathisches Ehlers-Danlos-Syndrom (mEDS)
- Parodontales Ehlers-Danlos-Syndrom (pEDS)
Ehlers-Danlos-Syndrom
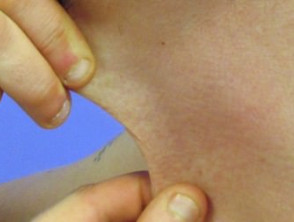
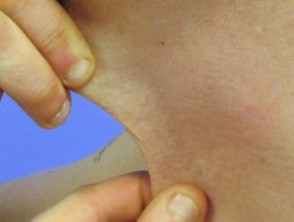
Hyperelastizität der Halshaut
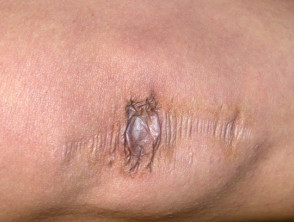
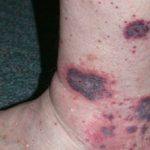
Umfangreiche Narbenbildung bei einem Patienten mit Ehlers-Danlos-Syndrom.
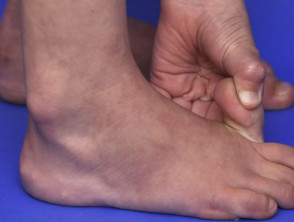
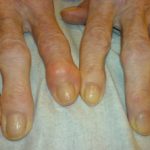
Hypermobile Gelenke
Weitere Bilder des Ehlers-Danlos-Syndroms.
Klassisches Ehlers-Danlos-Syndrom (cEDS)
Das Vererbungsmuster von cEDS ist autosomal Dominant.
- Die betroffenen Gene in cEDS sind COL5A1 (MIM130000), COL5A2, (MIM 130010) und COL1A1 Typ V und Typ I betreffen Kollagen.
- 50% der Patienten, bei denen cEDS diagnostiziert wurde, haben a neu Mutation (dh kein Elternteil ist betroffen).
- Der Schweregrad variiert erheblich, sogar innerhalb derselben Familie.
- Gefäß Zerbrechlichkeit kann eine spontane Dissektion oder eine mediane Ruptur umfassen Arterien.
- Patienten mit cEDS haben typischerweise eine ausgeprägte, erweiterte Hautüberdehnbarkeit atrophisch Haut- Narben und weit verbreitet Gelenkhypermobilität.
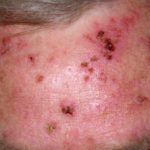
- Weitere Merkmale von cEDS sind Hämosiderin Erklärung (besonders auffällig an Schienbeinen und Handrücken), subkutan Sphäroide (Fettlappen, die nach Verlust der Blutversorgung verkalkt sind) und molluskoide Pseudotumoren (verdickte fleischige Haut Verletzungen/ Narben an Knien und Ellbogen).
- Es gibt keine Schlieren (Striae) in cEDS.
- typisches Elektron Mikroskopie Befunde sind „Kollagenblüten“ oder „Blumenkohl“ aufgrund einer abnormalen Fibrillogenese.
- Komplikationen von cEDS sind Herzklappen Missbildungen, Dilatation der Aorta und Aorta Präparation
Ehlers-Danlos-Syndrom vom klassischen Typ (clEDS)
Das Vererbungsmuster von clEDS ist autosomal rezessiv.
- Die betroffenen Gen in cLEDS ist TNXB Einfluss auf die Protein Tensin XB (MIM 606408)
- Patienten mit clEDS haben eine allgemeine Hypermobilität der Gelenke, eine samtweiche, überdehnbare Haut und schwere Blutergüsse.
- Bei cLEDS gibt es keine atrophischen Narben.
Herzklappen-Ehlers-Danlos-Syndrom (cvEDS)
Das Vererbungsmuster von cvEDS ist autosomal-rezessiv.
- Das betroffene Gen in cvEDS ist COL1A2 Beeinflussung des Kollagentyp-I-Proteins (MIM 225320)
- Patienten mit cvEDS weisen leichte Anzeichen von EDS auf, haben aber einen schweren Aortenfehler/eine Beeinträchtigung der Herzklappe, der einen chirurgischen Eingriff erfordert.
Vaskuläres Ehlers-Danlos-Syndrom (vEDS)
Das Vererbungsmuster von vEDS ist autosomal dominant.
- Die betroffenen Gene in vEDS sind COL3A1 Beeinflussung von Kollagen Typ III und COL1A1 Beeinflussung von Kollagen Typ 1 (MIM 130050)
- 50% der Patienten, bei denen vEDS diagnostiziert wurde, haben a de novo Mutation (d.h. kein Elternteil ist betroffen).
- das Median Die Lebenserwartung beträgt 50 Jahre.
- Die subtilen oder fehlenden klinischen Merkmale von vEDS bei einigen Patienten erschweren die Diagnose dieses Subtyps, wobei ein plötzlicher Tod im Alter zwischen 30 und 40 das einzige Merkmal ist.
- vEDS kann bereits bei der Geburt festgestellt werden, wenn ein Klumpfuß auffällt Deformität oder Hüftluxation.
- Kennzeichen von vEDS sind schwere, dünne Blutergüsse durchscheinend Charakteristisches Haut- und Gesichtsbild (straffe, eingefallene Wangen, schmale Lippen und Nase sowie hervorstehende Augen).
- Die Komplikationen von vEDS sind arteriell Dissektion (einschließlich der Aorta), Ruptur (einschließlich Darmruptur, wobei das Sigma die häufigste Stelle ist) und geburtshilfliche Komplikationen (Uterusruptur, massive postpartale Erkrankung). Blutung).
Hypermobiles Ehlers-Danlos-Syndrom (hEDS)
Das Vererbungsmuster von hEDS ist autosomal-dominant.
- Das betroffene Gen in hEDS (MIM 130020) ist unbekannt.
- Patienten mit hEDS sind nicht anfällig für lebensbedrohliche Komplikationen, aber ihre Auswirkungen können schwächend sein.
- Charakteristisch für hEDS sind eine generalisierte Hypermobilität der Gelenke, eine Hypermobilität der Wirbelsäule und häufige Gelenkluxationen oder -subluxationen (Schulter, Patella oder Kiefergelenk).
- Es gibt keine Molluscum-Pseudotumoren.
- Die Komplikationen von hEDS sind Dilatation der Aortenwurzel, autonom Funktionsstörung (Haltungsorthostatik Tachykardie Syndrom), schwer chronisch Gelenkschmerzen, Skolioseund verfrüht Arthrose.
Arthrochalasie-Ehlers-Danlos-Syndrom (aEDS)
Das Vererbungsmuster von aEDS ist autosomal-dominant.
- Die betroffenen Gene bei aEDS sind COL1A1 und COL1A2 Beeinflussung von Kollagen Typ I (MIM 130060).
- Wie das Wort schon sagt, bedeutet arthro- Artikulation und -chalasie bedeutet Entspannung; Die Mutation führt zu einem ungewöhnlich schwachen Kollagen.
- Patienten mit AEDS haben ein lebenslanges Risiko von gleichzeitig Luxation mehrerer großer Gelenke.
- Die Funktionen von aEDS sind Hypotonie, schwere generalisierte Gelenkhypermobilität, angeboren bilateral Hüftluxation und Osteopenie.
Dermatosparaxis Ehlers-Danlos-Syndrom (dEDS)
Das Vererbungsmuster von dEDS ist autosomal-rezessiv.
- Das betroffene Gen in dEDS ist ADAMTS2 das das ADAMTS-2-Protein (MIM 225410) betrifft.
- Sparaxis leitet sich von einem griechischen Wort ab, das „zerreißen“ bedeutet.
- Merkmale von dEDS sind schwere Hautbrüchigkeit, lose überflüssige Haut, verzögerter Verschluss der Fontanellen, blaue Farbe Lederhaut, viszeral Gebrechlichkeit und postnatale Wachstumsverzögerung, die zu Kleinwuchs führt.
- Es gibt hieroglyphische Merkmale Fibrillen' in Elektronenmikroskop der Haut.
Kyphoskoliotisches Ehlers-Danlos-Syndrom (kEDS)
Das Vererbungsmuster von kEDS ist autosomal-rezessiv.
- Die bei dEDS betroffenen Gene sind PLOD1 die das LH1-Protein (MIM 225400) beeinflussen, und FKBP14 Beeinflussung des FKBP22-Proteins (MIM 614557).
- Gen Mutationen zu einem Mangel an Lysylhydroxylase, einem Kollagenmodifikator, führen Enzym Wichtig für die Bildung von Querverbindungen zwischen Kollagentrimeren, um die Kollagenstärke zu erhöhen.
- kEDS ist durch einen frühen Beginn gekennzeichnet progressiv Kyphoskoliose, Hypotonie, grobmotorische Verzögerung, blaue Sklera mit erhöhtem Augeninnendruck, allgemeine Gelenkhypermobilität und leichte Blutergüsse.
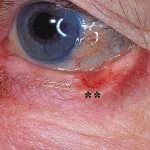
Brittle-Cornea-Syndrom (BCS)
Das Vererbungsmuster von BCS ist autosomal-rezessiv.
- Die bei dEDS betroffenen Gene sind PRDM5 Beeinflussung des Proteins PRDM5 (MIM 614170) und ZNF469 Beeinflussung von ZNF469 (MIM 229200).
- BCS-Patienten haben eine dünne und zerbrechliche Hornhaut, die das kann ausstanzen spontan oder nach Moll Trauma.
- Merkmale von BCS sind dünne Hornhaut, Keratokonus, blaue Sklera, Risiko einer Bulbusruptur, Netzhautablösung und hohe Kurzsichtigkeit (Myopie).
Ehlers-Danlos-spondylodysplastisches Syndrom (spEDS)
Das Vererbungsmuster von spEDS ist autosomal-rezessiv.
- Die betroffenen Gene in spEDS sind B4GALT7 das das Protein beta4GalT7 (MIM 130070) beeinflusst, B3GALT6 Beeinflussung von beta3GalT6 (MIM 615349) und SLC39A13 Auswirkungen auf ZIP13 (MIM 612350).
- Merkmale von spEDS sind Kleinwuchs, Hypotonie, Verkrümmung der Extremitäten, Osteopenie und überdehnbare, dünne und teigige Haut.
Ehlers-Danlos muskulokontrakturales Syndrom (mcEDS)
Das Vererbungsmuster von mcEDS ist autosomal-rezessiv.
- Die betroffenen Gene in mcEDS sind CHST14 Beeinflussung des D4ST1-Proteins (MIM 601776) und DSE Auswirkungen auf DSE (MIM 615539).
- Merkmale von mcEDS sind angeboren Kontrakturen dadurch Klumpfuß, wiederkehrend Luxationen, adduzierte (angezogene) Daumen in der Kindheit, überdehnbare Haut, leichte Blutergüsse, spröde Haut und atrophische Narben.
Myopathisches Ehlers-Danlos-Syndrom (mEDS)
Vererbungsmuster von mEDS können autosomal-dominant oder autosomal-rezessiv sein.
- Das betroffene Gen in mEDS ist COL12A1 Beeinflussung von Kollagen Typ XII (MIM 616471).
- Muskelschwäche, die im Säuglings- und Kindesalter vorhanden ist, verbessert sich tendenziell bis ins Erwachsenenalter, mit einer gewissen Verschlechterung nach dem 40.
Parodontales Ehlers-Danlos-Syndrom (pEDS)
Das Vererbungsmuster von pEDS ist autosomal-dominant.
- Die betroffenen Gene in pEDS sind C1R die das C1r-Protein beeinflussen und C1S Auswirkungen auf C1 (MIM 130080).
- Die Charakteristika von pEDS sind früh einsetzend schwerwiegend Parodontitis (rot geschwollenes Zahnfleisch, Zahnfleischrückgang, früher Zahnverlust), gelblicher Bluterguss pretibial Platten, Gelenkhypermobilität und überdehnbare Haut.