Clasificación Internacional de 2017 de los Síndromes de Ehlers-Danlos
Se hace referencia al Síndrome de Ehlers-Danlos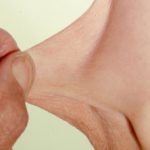 Comprendiendo el Síndrome de Ehlers-Danlos (SED): Causas y Características El Síndrome de Ehlers-Danlos (SED) constituye un grupo de trastornos hereditarios caracterizados por un defecto genético específico en la síntesis y estructura del colágeno o del tejido conectivo [síntesis] [1]. Esta alteración fundamental produce una tríada de manifestaciones clínicas principales: • Piel exhibiting fragilidad e hiperextensibilidad. • Articulaciones que presentan inestabilidad e hiperextensibilidad (hipermóviles). • Tejidos y vasos sanguíneos frágiles. Manifestaciones más para una descripción general del trastorno.
Comprendiendo el Síndrome de Ehlers-Danlos (SED): Causas y Características El Síndrome de Ehlers-Danlos (SED) constituye un grupo de trastornos hereditarios caracterizados por un defecto genético específico en la síntesis y estructura del colágeno o del tejido conectivo [síntesis] [1]. Esta alteración fundamental produce una tríada de manifestaciones clínicas principales: • Piel exhibiting fragilidad e hiperextensibilidad. • Articulaciones que presentan inestabilidad e hiperextensibilidad (hipermóviles). • Tejidos y vasos sanguíneos frágiles. Manifestaciones más para una descripción general del trastorno.
A continuación, se detallan los subtipos específicos o síndromes clasificados bajo el paraguas del Síndrome de Ehlers-Danlos (SED), según la clasificación [1,2]. Estos incluyen:
- Síndrome de Ehlers-Danlos clásico (cEDS)
- Síndrome de Ehlers-Danlos tipo clásico (clEDS)
- Síndrome de Ehlers-Danlos cardio-valvular (cvEDS)
- Síndrome de Ehlers-Danlos Vascular (vEDS)
- Síndrome de Ehlers-Danlos hipermóvil (hEDS)
- Síndrome de Ehlers-Danlos por artrocalasia (aEDS)
- Síndrome de Ehlers-Danlos por dermatosparaxis (dEDS)
- Síndrome de Ehlers-Danlos cifoescoliótico (kEDS)
- Síndrome de la córnea frágil (BCS)
- Síndrome de Ehlers-Danlos espondilodisplásico (spEDS)
- Síndrome de Ehlers-Danlos miocontractural (mcEDS)
- Síndrome de Ehlers-Danlos miopático (mEDS)
- Síndrome de Ehlers-Danlos periodontal (pEDS)
Imágenes Relacionadas con el Síndrome de Ehlers-Danlos
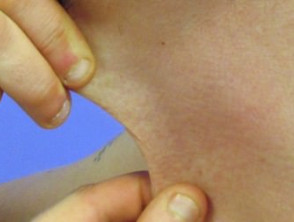
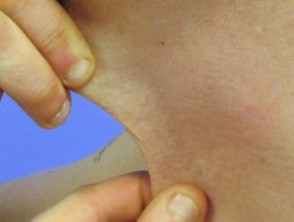
Hiperelasticidad de la piel del cuello
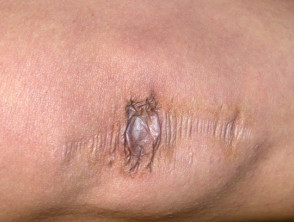
Amplia cicatrización en paciente con síndrome de Ehlers-Danlos.
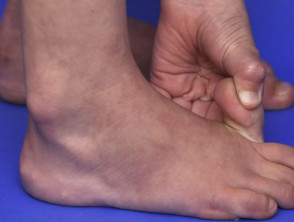
Articulaciones hipermóviles
Visualice más ejemplos del Síndrome de Ehlers-Danlos en nuestra galería.
Síndrome de Ehlers-Danlos clásico (cEDS)
El patrón de herencia del cEDS se clasifica como autosómico dominante, lo que significa que solo se necesita una copia del gen alterado para manifestar la condición.
- Los genes afectados en el cEDS son COL5A1 (MIM 130000), COL5A2 (MIM 130010) y COL1A1, los cuales inciden en la producción del colágeno tipo V y tipo I, esencialmente el colágeno estructural.
- Aproximadamente el 50% de los individuos diagnosticados con cEDS presentan una mutación de novo, es decir, una alteración genética no heredada de ninguno de los progenitores.
- La severidad clínica de esta condición muestra una gran variabilidad, incluso entre miembros de la misma familia.
- Puede presentarse fragilidad arterial, lo que conlleva riesgo de disección espontánea o ruptura de las arterias medianas.
- Típicamente, los pacientes con cEDS manifiestan una hiperextensibilidad cutánea notable, cicatrices cutáneas atróficas y anchas, y una hipermovilidad articular generalizada.
- Otras manifestaciones clínicas incluyen depósitos de hemosiderina (especialmente visibles en espinillas y dorso de las manos), formación de esferoides subcutáneos (nódulos grasos calcificados tras pérdida de irrigación sanguínea) y lesiones o cicatrices de aspecto pseudotumoral moluscoide (engrosamiento de la piel en rodillas y codos).
- Es importante destacar que las estrías
 ¿Qué son las estrías y sus tipos? Las estrías son fisuras o líneas finas que aparecen en la piel, resultado del desgarro del tejido subcutáneo provocado por el estiramiento rápido o excesivo de la misma. Aunque representan una condición muy común y generalmente no suponen un riesgo médico significativo, pueden generar preocupación estética en ciertas personas. Existen diversas denominaciones para referirse a las estrías, basadas en su apariencia, causa o más (estrías atróficas) generalmente no se observan en el cEDS.
¿Qué son las estrías y sus tipos? Las estrías son fisuras o líneas finas que aparecen en la piel, resultado del desgarro del tejido subcutáneo provocado por el estiramiento rápido o excesivo de la misma. Aunque representan una condición muy común y generalmente no suponen un riesgo médico significativo, pueden generar preocupación estética en ciertas personas. Existen diversas denominaciones para referirse a las estrías, basadas en su apariencia, causa o más (estrías atróficas) generalmente no se observan en el cEDS. - Los hallazgos en la microscopía electrónica son característicos, mostrando estructuras conocidas como «flores de colágeno» o «coliflores» debido a una formación defectuosa de las fibrillas.
- Las complicaciones potenciales del cEDS abarcan desde deformidades en las válvulas cardíacas hasta la dilatación aórtica.
Continuar explorando los detalles de cada síndrome ayuda a comprender mejor el espectro completo de los trastornos del tejido conectivo clasificados en 2017.
Tipos de Síndrome de Ehlers-Danlos (SED) y sus Características
La clasificación de los distintos tipos del Síndrome de Ehlers-Danlos (SED) es crucial para el diagnóstico y manejo, ya que cada subtipo presenta patrones de herencia, genes específicos y manifestaciones clínicas variadas, desde problemas articulares hasta graves implicaciones vasculares. A continuación, se detallan los subtipos más reconocidos.
- Un riesgo asociado es la disección aórtica.
Síndrome de Ehlers-Danlos de Tipo Clásico (clEDS)
El patrón de herencia que define al clEDS es autosómica recesiva. Este tipo se distingue por varias características cutáneas y articulares específicas.
- El gen afectado en el clEDS es TNXB, el cual codifica la proteína Tenascin XB (MIM 606408).
- Los pacientes experimentan hipermovilidad articular generalizada, una piel hiperextensible, suave y aterciopelada, y desarrollan hematomas con facilidad.
- Una característica distintiva es la ausencia de cicatrices atróficas en el clEDS.
Síndrome de Ehlers-Danlos Cardíaco-Valvular (cvEDS)
El cvEDS también sigue un patrón de herencia autosómico recesivo. Aunque los signos generales pueden ser leves, presenta un compromiso cardíaco significativo.
- El gen causal del cvEDS es COL1A2, que afecta a la proteína colágeno tipo I (MIM 225320).
- Aunque los pacientes pueden mostrar signos leves de EDS, sufren un defecto aórtico grave o compromiso de la válvula cardíaca que frecuentemente requiere intervención quirúrgica.
Síndrome de Ehlers-Danlos Vascular (vEDS)
El vEDS posee un patrón de herencia dominante autosómico. Este subtipo es conocido por sus complicaciones potencialmente fatales relacionadas con la fragilidad de los tejidos conectivos, especialmente en los vasos sanguíneos y órganos internos.
- Los genes primarios afectados son COL3A1, que afecta al colágeno tipo III, y COL1A1, que codifica el colágeno tipo I (MIM 130050).
- Aproximadamente el 50% de los individuos diagnosticados con vEDS presentan una mutación de novo, lo que implica que ninguno de los progenitores porta la mutación.
- La mediana esperanza de vida para los afectados se sitúa alrededor de los 50 años.
- Diagnosticar el vEDS puede ser complicado, ya que algunos pacientes presentan características clínicas sutiles o nulas, llevando a que la primera manifestación sea una muerte súbita entre los 30 y 40 años.
- El vEDS puede ser identificado al nacer si existe una marcada deformidad de pie zambo o una luxación de cadera congénita.
- Las características clínicas notables incluyen hematomas severos, piel delgada y translúcida, y un fenotipo facial característico (labios y nariz finos, mejillas hundidas y tensas, ojos prominentes).
- Las complicaciones graves del vEDS abarcan la disección arterial (incluida la aorta), la ruptura de órganos (siendo el colon sigmoide el sitio más habitual para la ruptura intestinal) y complicaciones obstétricas como la ruptura uterina o una hemorragia posparto masiva.
Síndrome de Ehlers-Danlos Hipermóvil (hEDS)
El hEDS tiene un patrón de herencia autosómico dominante. A diferencia de otros tipos, el gen específico responsable aún se encuentra desconocido (MIM 130020).
- Los pacientes con hEDS generalmente no enfrentan complicaciones potencialmente mortales, sin embargo, el impacto de sus síntomas puede ser altamente debilitante.
- Las manifestaciones principales del hEDS incluyen hipermovilidad articular generalizada, extensa hipermovilidad espinal y una propensión a luxaciones o subluxaciones frecuentes de articulaciones clave como el hombro, la rótula o la articulación temporomandibular.
- No se observan pseudotumores de molusco en este subtipo.
- Las complicaciones asociadas al hEDS incluyen dilatación de la raíz aórtica, disfunción autonómica (manifestada como síndrome de taquicardia postural ortostática), dolor articular crónico severo, escoliosis y osteoartritis prematura.
Síndrome de Ehlers-Danlos por Artrocalasia (aEDS)
El aEDS se hereda de forma autosómica dominante. Su nomenclatura, que combina «artro» (articulación) y «calasia» (relajación), resalta su afectación principal.
- Los genes implicados son COL1A1 y COL1A2, ambos afectando al colágeno de tipo I (MIM 130060).
- La mutación resultante produce un colágeno débil y anómalo.
- Los individuos con aEDS enfrentan un riesgo significativo a lo largo de su vida de sufrir la concurrente dislocación de múltiples articulaciones mayores.
- Las características clínicas del aEDS incluyen hipotonía, hiperlaxitud articular generalizada severa, luxación de cadera congénito y bilateral, además de osteopenia.
Comprender estas diferencias genéticas y sintomáticas es fundamental para proveer la atención médica especializada que cada variante del SED requiere. La investigación continúa buscando terapias dirigidas para mitigar tanto las complicaciones vasculares como el impacto crónico en la calidad de vida de los pacientes.
Tipos Raros del Síndrome de Ehlers-Danlos (SED)
Síndrome de Ehlers-Danlos Dermatosparáxico (dEDS)
El patrón de herencia característico del SED Dermatosparáxico es autosómico recesivo.
- El gen implicado en el dEDS es ADAMTS2, que codifica la proteína ADAMTS-2 (MIM 225410).
- El término «sparaxis» proviene de una palabra griega que significa «rasgarse».
- Las manifestaciones clínicas del dEDS incluyen fragilidad cutánea intensa, redundancia y laxitud de la piel, cierre tardío de las fontanelas, coloración azulada de las escleróticas, fragilidad visceral y retraso en el crecimiento posnatal, lo que resulta en baja estatura.
- Se observan en el microscopio electrónico de la piel características de fibrillas con apariencia jeroglífica.
Síndrome de Ehlers-Danlos Cifoscoliótico (kEDS)
El patrón de herencia del kEDS también es autosómico recesivo.
- Los genes afectados son PLOD1, que impacta la proteína LH1 (MIM 225400), y FKBP14, que afecta a la proteína FKBP22 (MIM 614557).
- Las mutaciones génicas provocan un déficit de lisilhidroxilasa, una enzima modificadora del colágeno esencial para formar enlaces cruzados entre los trímeros de colágeno, lo que aumenta la resistencia estructural del mismo.
- El kEDS se distingue por una cifoescoliosis progresiva de inicio temprano, hipotonía, retraso en la adquisición de la motricidad gruesa, escleróticas azules asociadas a presión intraocular elevada, hipermovilidad articular generalizada y tendencia al hematoma fácil.
Síndrome de la Córnea Frágil (BCS)
El patrón de herencia del BCS se transmite de forma autosómica recesiva.
- Los genes asociados son PRDM5, que afecta a la proteína PRDM5 (MIM 614170), y ZNF469, que afecta a ZNF469 (MIM 229200).
- Los pacientes con BCS presentan una córnea delgada y sumamente frágil, susceptible a la perforación espontánea o tras un trauma menor.
- Las características predominantes incluyen córnea delgada, queratocono, esclerótica azul, riesgo elevado de rotura ocular, desprendimiento de retina y miopía alta (refractiva).
Síndrome de Ehlers-Danlos Espondilodisplásico (spEDS)
El patrón de herencia del spEDS es autosómico recesivo.
- Los genes implicados son B4GALT7, afectando a la proteína beta4GalT7 (MIM 130070); B3GALT6, que afecta a beta3GalT6 (MIM 615349); y SLC39A13, que afecta a ZIP13 (MIM 612350).
- Las manifestaciones clínicas del spEDS abarcan baja estatura, hipotonía, arqueamiento de las extremidades, osteopenia y una piel hiperextensible, fina y con aspecto pastoso.
Síndrome de Ehlers-Danlos Musculocontractural (mcEDS)
El patrón de herencia del mcEDS es autosómico recesivo.
- Los genes asociados son CHST14, que codifica la proteína D4ST1 (MIM 601776), y DSE, que afecta a DSE (MIM 615539).
- Las características del mcEDS incluyen contracturas congénitas que resultan en pie equinovaro (zambo), dislocaciones recurrentes, pulgares en aducción (posición cerrada) durante la infancia, piel hiperextensible, hematomas frecuentes y cicatrización atrófica.
Síndrome de Ehlers-Danlos Miopático (mEDS)
El patrón de herencia del mEDS puede manifestarse como autosómico dominante o autosómico recesivo.
- El gen afectado en el mEDS es COL12A1, que codifica el colágeno tipo XII (MIM 616471).
- La debilidad muscular que se presenta en la infancia y niñez tiende a mejorar con el avance hacia la edad adulta, aunque se puede observar un cierto deterioro funcional después de los 40 años.
Síndrome de Ehlers-Danlos Periodontal (pEDS)
El patrón de herencia del pEDS es autosómico dominante.
- Los genes implicated son C1R, que afecta a la proteína C1r, y C1S, que afecta a C1 (MIM 130080).
- Las manifestaciones del pEDS incluyen una periodontitis grave de inicio temprano (caracterizada por encías inflamadas y rojas, recesión gingival y pérdida dental prematura), placas cutáneas amarillentas pretibiales, hipermovilidad articular y piel hiperextensible.
Comprender las distinciones entre estos subtipos raros del Síndrome de Ehlers-Danlos es fundamental para establecer un diagnóstico preciso y definir el manejo clínico más adecuado para cada paciente, dada su base genética heterogénea.


