Was ist es Dyskeratose angeboren?
Dyskeratosis congenita ist auch als Zinsser-Engman-Cole bekannt Syndrom. Es ist eine Gruppe von genetisch Krankheiten, die am häufigsten Manifest mit mukokutan Anzeichen, Versagen des Knochenmarks und / oder der Lunge oder Leber Fibrose.
Der Schweregrad, das Erkrankungsalter und die Organbeteiligung sind selbst innerhalb einzelner Familien sehr unterschiedlich. Es ist jetzt bekannt, dass die verschiedenen klinischen Merkmale auf 'Telomer Verkürzung '(instabil Chromosomen), eine der Ursachen für vorzeitiges Altern. Seit die genetischen Veränderungen identifiziert wurden, wurde festgestellt, dass eine Reihe anderer Zustände auf Veränderungen derselben zurückzuführen sind Gene.
Wer bekommt Dyskeratosis congenita?
Dyskeratosis congenita wird vererbt und tritt normalerweise in der Kindheit auf. Varianten umfassen:
- Autosomal Dominante Dyskeratosis congenita (die Gen kommt von einem Elternteil): MIM * # 127550
- Autosomal rezessiv Dyskeratosis congenita (ein Gen stammt von jedem Elternteil): MIM # 224230
- X-chromosomale Dyskeratosis congenitaChromosom (betrifft nur Männer) MIM # 305000
- Sporadische Formen der Dyskeratosis congenita.
* MIM ist eine Abkürzung für Mendelsche Vererbung im Menschen
Autosomal dominant Formen der Dyskeratosis congenita zeigen eine „genetische Antizipation“, bei der der Vater eines betroffenen Kindes die Mutation aber es zeigt keine Anzeichen der Erkrankung und jede nachfolgende Generation entwickelt früher Anzeichen einer Krankheit.
Die Schwere der Erkrankung hängt von der Länge der Telomere ab: Je kürzer die Telomere Je schwerer die Krankheit, und dies kann oft mit der Art der Vererbung zusammenhängen. Im Allgemeinen ist die X-verknüpfte Form die schwerste, die autosomal dominante Form die mildeste und der autosomal rezessive Typ kann irgendwo zwischen den beiden Extremen liegen.
Klinische Merkmale der Dyskeratosis congenita
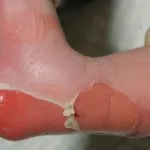
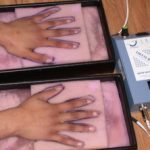
Die klassische mukokutane Triade der Dyskeratosis congenita wurde ursprünglich durch die folgenden drei mukokutanen Merkmale definiert:
- EIN Dystrophie
- Längsrichtung Rille
- Verschwendung von Nagel
- Coilonychia (dünne, löffelförmige Nägel)
- Abnormale Haut Pigmentierung
- Retikulieren (Spitze) Hyperpigmentierung Hautfalten, beschrieben als "schmutziger Hals" (> 90%)
- Orale Leukoplakie
- Erscheint später in der Kindheit.
- Weiße Flecken im Mund
Die ersten beiden Merkmale treten normalerweise im Alter von 10 Jahren auf.
Andere Hautveränderungen können sein:
- Früh Haar Ergrauung oder Haarausfall
- Spärliche Wimpern
- Nagel streifend
- Hyperhidrose (starkes Schwitzen)
- Schuppig Zelle Karzinome (SCC), die häufig im Teenageralter oder in den Zwanzigern auftritt (50% im Alter von 40 Jahren). SCC wird oft in gefunden Schleimhaut Websites, die mündliche enthalten Krebs
Knochenmarkversagen entwickelt sich im Alter von 30 Jahren in 80-90%.
Neben Hautkrebs treten bei diesem Syndrom normalerweise viele andere Krebsarten auf Entwicklung In den zwanziger Jahren:
- Mund: Mundkrebs, insbesondere SCC der Zunge
- Atemwege - Karzinome von Larynx oder Bronchus
- Verdauungstrakt - Karzinome von Speiseröhre, Dickdarm, Bauchspeicheldrüse und anorektal
- Knochenmark - Leukämien, speziell, akut myeloisch Leukämie es ist 200-mal häufiger als in der allgemeinen Bevölkerung.
- Lymphome
Die neue Klassifikation der Dyskeratosis congenita
Mit der Identifizierung der Genetik Mutationen Dyskeratosis congenita, andere zuvor nicht verwandt Demonstrationen jetzt haben sie verlinkt. Eine vorgeschlagene Änderung der Definition ist:
- Eine oder mehrere Eigenschaften der klassischen mukokutanen Triade
- Knochenmarkversagen wie aplastisch Anämie
- Zwei oder mehr der anderen inneren Veränderungen, von denen bekannt ist, dass sie bei Dyskeratosis congenita auftreten:
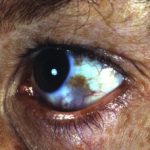
- Augen - übermäßige Tränen
- Nervensystem - Lernschwierigkeiten, Entwicklung/ geistige Behinderung, Ataxie (Instabilität), unterentwickeltes Kleinhirn, Mikrozephalie (kleines Gehirn), Taubheit
- Lungen - Lungen Fibrose (Narbenbildung), abnormal Blutgefäße
- Knochen: Osteoporose (dünne Knochen), aseptisch Nekrose (Knochentod), Skoliose (krumme Wirbelsäule), Kleinwuchs
- Zähne: ausgedehnte Karies und Verlust, verringertes Wurzel / Kronen-Verhältnis, leichter Taurodontismus (Vergrößerung der Zähne und der Pulpakammer)
- Verdauungssystem - Fibrose / Leberversagen, Speiseröhre Stenose (Verengung), peptisch Geschwür
- Urogenital System - Hypogonadismus (kleine Hoden), Hoden, Phimose (enge Vorhaut), Harnröhre Stenose (Verengung)
Kombinationen von klinischen Merkmalen, Erkrankungsalter und Schweregrad variieren je nach Mutation und innerhalb der Familien.
Derzeit wurde gezeigt, dass andere Erkrankungen die gleichen genetischen Mutationen aufweisen wie die klassische Dyskeratosis congenita. Diese schließen ein:
- Hoyeraal-Hreidarsson-Syndrom: sehr kleine Statur, Knochenmarkversagen, Immunschwäche, unterentwickeltes Kleinhirn
- idiopathisch aplastische Anämie (Anämie durch Knochenmarkversagen)
- Myelodysplasie (Versagen des Knochenmarks)
- idiopathische Lungenfibrose
- paroxysmale nächtliche Hämoglobinurie
- Revesz-Syndrom - bilateral exsudative Retinopathie (Augenkrankheit), Knochenmarkshypoplasie (Insuffizienz), Nageldystrophie, feines Haar, Kleinhirnhypoplasie, Wachstumsverzögerung
Wie wird Dyskeratosis congenita diagnostiziert?
Die Diagnose einer Dyskeratosis congenita basiert auf der obigen Definition.
Bisher wurden genetische Mutationen nur in etwa 50%-Fällen identifiziert.
Dyskeratosis congenita kann durch Fluss unterschieden werdenFISCH Analyse aufgrund sehr kurzer Telomere im Vergleich zu alten Kontrolle S..
Es ist wichtig, die Diagnose bei Knochenmarkversagen oder Lungenfibrose, bei denen keine andere Ursache festgestellt wurde, bei oraler Leukoplakie bei jungen Menschen ohne Rauchanamnese und bei Krebserkrankungen im Frühstadium zu berücksichtigen.
Was ist die Behandlung für Dyskeratosis congenita?
Derzeit gibt es keine Heilung für Dyskeratosis congenita. Die Behandlung zielt auf die Aufrechterhaltung der Knochenmarkfunktion ab, da dies die häufigste Todesursache ist:
- Oxymetholon - Ein anaboles Steroid, das die Knochenmarkfunktion bei zwei Dritteln der Patienten über mehrere Jahre hinweg unterstützt.
- Hämatopoetisch Wachstumsfaktoren - Erythropoetin, Granulozyt Makrophagen koloniestimulierender Faktor und Granulozytenkolonie stimulierender Faktor.
- Knochenmark Transplantation - Dies war bei Dyskeratosis congenita aufgrund der schnellen ausnahmslos tödlich Entwicklung von Leberversagen nach der Transplantation oder Lungenfibrose. Es wurde nun erkannt, dass dies auf eine Intoleranz vor der Transplantation zurückzuführen ist. Chemotherapie mich Strahlentherapie. Knochenmarktransplantationen wurden bei einer kleinen Anzahl von Patienten mit Dyskeratosis congenita mit weniger aggressiver Therapie vor der Transplantation erfolgreich durchgeführt.
Dyskeratosis congenita scheint zwar eine ideale Voraussetzung für die Gentherapie zu sein, tut dies jedoch nicht Fortschritt wurde bisher in diese Richtung getan.
Genetische Beratung ist wichtig für die Planung zukünftiger Schwangerschaften. Die pränatale Diagnose wurde erfolgreich durchgeführt.
Was ist das Ergebnis von Dyskeratosis congenita?
Der Schweregrad und das Alter beim Tod sind sehr unterschiedlich. Einige Formen sind milder mit einem Überleben von vierzig; andere sind in der Kindheit tödlich. Die Haupttodesursachen sind Knochenmarkversagen bei einem 75-80% (aufgrund erhöhter Anfälligkeit zu Infektion oder Blutung), Lungenfibrose bei 10-15% oder Krebs bei 10%.
Wie entsteht eine angeborene Dyskeratose?
Das Telomer ist eine Region von repetitiv DNA das bildet eine Schicht am Ende eines Chromosoms in normalen Zellen. Schützt das Ende des Chromosoms und sorgt für dessen Stabilität. Die normale Zelle teilt sich während ihres Lebens etwa 50 Mal. Das Telomer verkürzt sich mit jeder Zellteilung. Letztendlich führt die Verkürzung der Telomere zu einer beschädigten DNA, die sich nicht mehr teilen kann. Daher ist eine Verkürzung der Telomere damit verbunden Mobiltelefon Altern.
EIN Enzym Die sogenannte Telomerase verhindert die Verkürzung der Telomere. Es wird von einigen Stammzellen und fast allen Krebszellen ausgeschieden, so dass sie sich weiter teilen können.
Derzeit wurden Mutationen in sechs kodierenden Genen identifiziert Protein an der Wartung von Telomeren beteiligt.
- Die meisten Mutationen betreffen die Telomerase. Ohne eine wirksame Telomerase verkürzt sich die Länge der Telomere mit jeder Zellteilung und wird schnell so kurz, dass die Zelle viel früher als normal stirbt. Dies ist am wichtigsten bei Zellen, die sich stark teilen, wie Knochenmark und Haut.
- Andere Mutationen beeinflussen die Protein "Schutz", der Telomere vor Schäden durch DNA-Reparaturmechanismen schützt.
| Art der Dyskeratosis congenita | Genetische Mutation |
|---|---|
| Autosomal dominant |
|
| Verbunden mit dem X-Chromosom |
|
| Autosomal rezessiv |
|
| Hoyeraal-Hreidarsson-Syndrom Revesz-Syndrom Einige Fälle von idiopathischer aplastischer Anämie |
|