Comprendiendo la Disqueratosis Congénita: Causas, Herencia y Manifestaciones Clínicas
¿Qué es la Disqueratosis Congénita?
La Disqueratosis Congénita (DC), también denominada Síndrome de Zinsser-Engman-Cole, constituye un conjunto de genéticas enfermedades. Estas se manifiestan típicamente a través de signos mucocutáneos, acompañados frecuentemente de insuficiencia de la médula ósea y/o desarrollo de fibrosis pulmonar o hepática.
Existe una marcada variabilidad en la severidad, la edad de inicio y la afectación orgánica, incluso dentro de los miembros de una misma familia. Hoy se comprende que las distintas características clínicas son resultado del «acortamiento de los telómeros» (lo que implica cromosomas inestables), siendo esta una de las causas subyacentes del envejecimiento prematuro. Desde que se identificaron las alteraciones genéticas específicas, se ha descubierto que varias otras afecciones comparten mutaciones en los mismos genes.
¿Quién padece Disqueratosis Congénita y Cómo se Hereda?
La Disqueratosis Congénita se hereda y su presentación clínica ocurre generalmente durante la niñez. Las variantes hereditarias principales incluyen:
- Disqueratosis Congénita autosómica dominante (la alteración del gen proviene de uno de los progenitores): MIM *#127550
- Disqueratosis Congénita autosómica recesiva (se hereda una copia del gen de cada progenitor): MIM #224230
- Disqueratosis Congénita ligada al cromosoma X (afecta predominantemente a varones): MIM #305000
- Formas esporádicas de Disqueratosis Congénita (sin herencia familiar clara).
* MIM es la abreviatura de Herencia Mendeliana en el Hombre.
Las formas autosómicas dominantes de la disqueratosis congénita exhiben un fenómeno conocido como «anticipación genética». Esto significa que el padre del niño afectado puede portar la mutación sin mostrar signos evidentes de la condición, pero cada generación subsiguiente experimenta la aparición de los signos de la enfermedad a una edad más temprana y, potencialmente, con mayor gravedad.
La severidad de la enfermedad está directamente correlacionada con la longitud de los telómeros: cuanto más cortos estén, más grave será el cuadro clínico. Esto a menudo se correlaciona con el modo de herencia. Típicamente, la forma ligada al cromosoma X se considera la más grave, la autosómica dominante la más leve, y la autosómica recesiva puede situarse en algún punto intermedio entre ambos extremos.
Características Clínicas: La Tríada Clásica de la Disqueratosis Congénita
La tríada mucocutánea clásica utilizada para definir la Disqueratosis Congénita se compone originalmente de las siguientes tres manifestaciones cutáneas y mucosas:
-
Distrofia ungueal
- Surco longitudinal
- Pérdida de uñas (aniquilación)
- Coiloniquia (deformación de las uñas, haciéndolas cóncavas o en forma de cuchara)
-
Pigmentación cutánea anómala
- Hiperpigmentación reticular (en forma de encaje, similar a una red) en los pliegues cutáneos, frecuentemente descrita como 'cuello sucio' (presente en más del 90% de los casos).
-
Leucoplasia oralEntendiendo la Leucoplasia Oral: Causas, Diagnóstico y Tratamiento La leucoplasia oral se define como la presencia de una mancha o placa de color blanco en la mucosa bucal que no puede ser clasificada clínica o patológicamente como ninguna otra enfermedad conocida. Posibles Causas y Factores Desencadenantes de la Leucoplasia OralLa aparición de leucoplasia oral puede estar relacionada con diversas condiciones subyacentes o hábitos. Entre las causas potenciales se incluyen: • más
- Generalmente se manifiesta más tardíamente en la infancia.
- Consiste en la aparición de manchas blancas dentro de la cavidad bucal.
Es importante notar que los dos primeros componentes de esta tríada, la distrofia ungueal y la alteración de la pigmentación, suelen ser los primeros signos que aparecen, generalmente antes de los 10 años de edad.
Adicionalmente, existen otros hallazgos clínicos sistémicos...
Las alteraciones cutáneas asociadas a esta afección pueden incluir:
- Inicio temprano de canas o pérdida de cabello.
- Presencia de pestañas escasas.
- Desarrollo de estrías
 ¿Qué son las estrías y sus tipos? Las estrías son fisuras o líneas finas que aparecen en la piel, resultado del desgarro del tejido subcutáneo provocado por el estiramiento rápido o excesivo de la misma. Aunque representan una condición muy común y generalmente no suponen un riesgo médico significativo, pueden generar preocupación estética en ciertas personas. Existen diversas denominaciones para referirse a las estrías, basadas en su apariencia, causa o más en las uñas.
¿Qué son las estrías y sus tipos? Las estrías son fisuras o líneas finas que aparecen en la piel, resultado del desgarro del tejido subcutáneo provocado por el estiramiento rápido o excesivo de la misma. Aunque representan una condición muy común y generalmente no suponen un riesgo médico significativo, pueden generar preocupación estética en ciertas personas. Existen diversas denominaciones para referirse a las estrías, basadas en su apariencia, causa o más en las uñas. - Hiperhidrosis
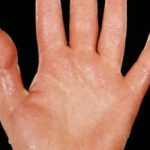 Comprendiendo la Hiperhidrosis: Síntomas, Causas y Población Afectada La hiperhidrosis se define como la sudoración excesiva e incontrolable. Este fenómeno va más allá de la transpiración normal necesaria para regular la temperatura corporal. El sudor es esencialmente una solución acuosa producida por las glándulas sudoríparas ecrinas. Estas glándulas se distribuyen por toda la superficie corporal, aunque se concentran significativamente en las palmas de las manos y las plantas de los más (sudoración excesiva).
Comprendiendo la Hiperhidrosis: Síntomas, Causas y Población Afectada La hiperhidrosis se define como la sudoración excesiva e incontrolable. Este fenómeno va más allá de la transpiración normal necesaria para regular la temperatura corporal. El sudor es esencialmente una solución acuosa producida por las glándulas sudoríparas ecrinas. Estas glándulas se distribuyen por toda la superficie corporal, aunque se concentran significativamente en las palmas de las manos y las plantas de los más (sudoración excesiva). - Aparición de carcinomas de células escamosas (SCC), que frecuentemente se manifiesta en la adolescencia o los veinte años (el 50% de los casos se presenta a los 40 años). El SCC se localiza a menudo en sitios de mucosa, como el cáncer oral.
Además, la insuficiencia de la médula ósea suele desarrollarse a la edad de 30 años en el 80-90% de los pacientes.
De forma paralela al cáncer de piel Comprendiendo el Cáncer de Piel Las neoplasias malignas cutáneas nacen de la proliferación celular descontrolada de cualquier tipo de célula de la piel. Este crecimiento anómalo difiere significativamente de la regeneración cutánea habitual, donde la replicación celular se mantiene rigurosamente regulada. Cada variante específica de cáncer cutáneo manifiesta características clínicas y patológicas propias. Las tres formas más prevalentes y comunes de malignidad en la piel incluyen: • Carcinoma de Células más, este síndrome se caracteriza por una alta incidencia de otros tipos de cáncer, los cuales tienden a desarrollarse en la veintena:
Comprendiendo el Cáncer de Piel Las neoplasias malignas cutáneas nacen de la proliferación celular descontrolada de cualquier tipo de célula de la piel. Este crecimiento anómalo difiere significativamente de la regeneración cutánea habitual, donde la replicación celular se mantiene rigurosamente regulada. Cada variante específica de cáncer cutáneo manifiesta características clínicas y patológicas propias. Las tres formas más prevalentes y comunes de malignidad en la piel incluyen: • Carcinoma de Células más, este síndrome se caracteriza por una alta incidencia de otros tipos de cáncer, los cuales tienden a desarrollarse en la veintena:
- Boca: Cáncer oralGuía Completa sobre el Cáncer Oral: Causas, Riesgos y Detección Temprana El espectro del cáncer oral abarca diversas tipologías, si bien el subtipo predominante es el carcinoma de células escamosas, que constituye más del 90% de todos los diagnósticos. A escala mundial, esta enfermedad se clasifica consistentemente entre las diez neoplasias más comunes. Aunque afecta a poblaciones diversas, su prevalencia se dispara en países en vías de desarrollo, siendo particularmente más, principalmente SCC de la lengua.
- Vías respiratorias: Carcinomas de laringe o bronquios.
- Tracto digestivo: Carcinomas de esófago, colon, páncreas y región anorrectal.
- Médula ósea: Leucemias; en particular, la leucemia mieloide aguda es 200 veces más prevalente que en la población general.
- Linfomas.
Nueva Clasificación de la Disqueratosis Congénita
Con el descubrimiento de las mutaciones genéticas causantes de la disqueratosis congénita, diversas manifestaciones previamente no relacionadas han sido asociadas a la enfermedad. Una propuesta de modificación en la definición sugiere:
- Uno o más de los componentes de la tríada mucocutánea clásica.
- Insuficiencia de la médula ósea, tal como la anemia aplásica.
- Dos o más de los siguientes cambios internos que se conocen por ocurrir en la disqueratosis congénita:
- Ojos: Producción excesiva de lágrimas.
- Sistema nervioso: Dificultades de aprendizaje, retraso del desarrollo o mental, ataxia (falta de estabilidad), subdesarrollo del cerebelo, microcefalia (cabeza pequeña), y sordera.
- Pulmones: Fibrosis pulmonar (cicatrización) y presencia de vasos sanguíneos anómalos.
- Huesos: Osteoporosis (huesos debilitados), necrosis aséptica (muerte de tejido óseo), escoliosis (curvatura de la columna), y baja estatura.
- Dientes: Caries extensas y pérdida dental, reducción de la proporción raíz/corona, y leve taurodontismo (aumento del tamaño de la cámara pulpar).
- Sistema digestivo: Fibrosis o insuficiencia hepática, estenosis esofágica (estrechamiento), y úlcera péptica.
- Sistema genitourinario: Hipogonadismo (testículos reducidos), testículos no descendidos, fimosis (prepucio constreñido) y estenosis uretral (estrechamiento).
La combinación de características clínicas, la edad de inicio y la severidad del cuadro se modifican según las diferentes mutaciones y varían entre distintos miembros de la misma familia.
Actualmente, se ha confirmado que otras patologías comparten las mismas bases genéticas que la disqueratosis congénita clásica. Entre ellas se encuentran:
- Síndrome de Hoyeraal-Hreidarsson: Implicando estatura muy baja, insuficiencia medular, inmunodeficienciaComprensión de la Inmunodeficiencia: Causas, Tipos y Consecuencias ¿Qué Provoca la Inmunodeficiencia y Cómo se Manifiesta? La inmunodeficiencia surge de un funcionamiento inadecuado del sistema inmunológico. Este estado puede ser desencadenado por una amplia variedad de factores, incluyendo síndromes hereditarios, infecciones, uso de ciertos medicamentos, condiciones médicas subyacentes, el embarazo, el proceso natural de envejecimiento y muchas otras causas. La gravedad de esta alteración es variable, lo que da lugar más y subdesarrollo cerebeloso.
- Anemia aplásica idiopática (originada por fallo medular).
- Mielodisplasia (insuficiencia de la médula ósea).
- Fibrosis pulmonar idiopática.
- Hemoglobinuria paroxística nocturna.
- Síndrome de Revesz: Caracterizado por retinopatía exudativa bilateral (problemas oculares), hipoplasia medular (insuficiencia), distrofia ungueal, cabello fino, hipoplasia cerebelosa y retraso en el crecimiento.
Diagnóstico de la Disqueratosis Congénita
El diagnóstico de la disqueratosis congénita se establece basándose en la definición clínica revisada anteriormente.
Es importante señalar que, hasta la fecha, las mutaciones genéticas específicas solo han sido identificadas en cerca del 50% de los casos reportados.
Puede ser factible diferenciar la disqueratosis congénita mediante el estudio de flujo
El PESCADO muestra un análisis caracterizado por telómeros significativamente más cortos en comparación con los sujetos de control S.
Es crucial considerar este diagnóstico en escenarios como la insuficiencia de la médula ósea o la fibrosis pulmonar sin una etiología identificable, en el caso de leucoplasia oral en pacientes jóvenes sin historial de tabaquismo, y en la aparición temprana de cánceres.
Tratamientos Disponibles para la Disqueratosis Congénita
Actualmente, no existe una cura definitiva para la disqueratosis congénita. El manejo clínico se enfoca principalmente en sustentar la funcionalidad de la médula ósea, dado que esta es la principal vía de mortalidad:
- Oximetolona: Este esteroide anabólico puede favorecer la función medular en aproximadamente dos tercios de los pacientes, manteniéndola activa durante varios años.
- Factores de crecimiento hematopoyéticos: Incluyen la eritropoyetina, el factor estimulante de colonias de granulocito macrófago y el factor estimulante de colonias de granulocitos.
- Trasplante de médula ósea: Históricamente, este procedimiento resultaba fatal debido al rápido desarrollo de fibrosis pulmonar o insuficiencia hepática postrasplante. Hoy se entiende que esto se debía a la intolerancia a la quimioterapia y/o radioterapia pretrasplante. Se han logrado trasplantes exitosos en casos seleccionados utilizando regímenes previos menos agresivos.
Aunque la disqueratosis congénita parece ser una afección ideal para la terapia génica, hasta la fecha no se ha logrado un Progreso significativo en esa dirección.
El asesoramiento genético es fundamental para la planificación familiar futura. Además, se han realizado diagnósticos prenatales exitosos.
Pronóstico y Supervivencia en la Disqueratosis Congénita
El nivel de severidad y la edad hasta el fallecimiento varían considerablemente. Algunas presentaciones son más leves, permitiendo la supervivencia hasta los cuarenta años, mientras que otras resultan fatales durante la infancia. Las causas primarias de deceso son la insuficiencia medular (en un 75-80% de los casos, motivada por un aumento de la susceptibilidad a la infección o hemorragia), la fibrosis pulmonar (10-15%), o el desarrollo de cáncer (10%).
Comprendiendo la Etiología de la Disqueratosis Congénita
El telómero es una estructura formada por secuencias de repetitivo ADN que corona cada extremo de un cromosoma en las células normales. Su función es proteger el extremo cromosómico y garantizar su estabilidad. Una célula normal típicamente se divide cerca de 50 veces durante su ciclo vital, y el telómero experimenta acortamiento con cada división. Eventualmente, este acortamiento resulta en ADN dañado que ya no puede replicarse; por ello, el acortamiento telomérico está intrínsecamente ligado al celular envejecimiento.
Una enzima conocida como telomerasa actúa previniendo este acortamiento telomérico. Esta enzima es secretada por ciertas células madre y por la vasta mayoría de las células cancerosas, lo que les confiere la capacidad de división continua.
Hasta ahora, se han identificado mutaciones en seis genes que codifican proteinas esenciales para la preservación de los telómeros.
- La mayoría de las mutaciones impactan la telomerasa. Sin una telomerasa funcional, la longitud telomérica disminuye con cada ciclo celular y rápidamente alcanza un punto crítico que induce la muerte celular prematura. Este efecto es especialmente notable en células con alta tasa de división, como las de la médula ósea y la piel.
- Otras mutaciones afectan a la 'protección de refugio' (proteína 'shelterin') que resguarda los telómeros de ser blanco de los sistemas de reparación del ADN.
| Tipo de Disqueratosis Congénita | Mutación Genética Asociada |
|---|---|
| Dominante autosómico |
|
| Ligado al cromosoma X |
|
| Autosómica recesiva |
|
| Síndrome de Hoyeraal-Hreidarsson Síndrome de Revesz Algunos casos de anemia aplásica idiopática |
|


