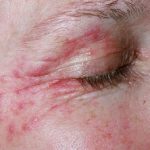
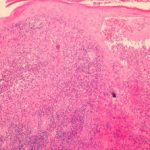
Was ist es Familie Mittelmeer- Fieber?
Das familiäre Mittelmeerfieber ist erblich autoinflammatorisch Syndrom charakterisiert durch wiederkehrend kurze Episoden von hohem Fieber verbunden mit Bauchschmerzen, Entzündung von Gelenken und anderen Körperteilen und der Haut Eruption. Wenn nicht behandelt, Amyloidose es entwickelt sich häufig und kann einen tödlichen Ausgang haben. Das familiäre Mittelmeerfieber ist das häufigste der periodischen Fiebersyndrome.
Beim familiären Mittelmeerfieber Typ 2 ist Amyloidose das charakteristische Merkmal ohne Vorgeschichte fieberhaft Episoden.
Wer bekommt familiäres Mittelmeerfieber und warum?
Das familiäre Mittelmeerfieber betrifft in erster Linie bestimmte Rassengruppen, die aus dem östlichen Mittelmeerraum stammen. Diese beinhalten:
- Araber
- Armenier
- Italiener (weiche Form)
- Juden
- Türken.
In diesen Gruppen ist 1 von 25 bis 1 von 2.000 Personen betroffen. Im Vergleich dazu ist bei Westeuropäern die Vorherrschaft es ist 2,5 pro 100.000 (1 in 40.000) Menschen. das Träger die Rate kann in einigen Gruppen bis zu 1 von 3 betragen. Obwohl früher angenommen wurde, dass es nur sephardische Juden betrifft, wurde kürzlich erkannt, dass aschkenasische Juden an einer milden Form des Syndroms leiden. Zwei Drittel der Patienten entwickeln Symptome vor 5 Jahren und 90% bei 20 Jahren.
Das familiäre Mittelmeerfieber wird in der Regel als a autosomal rezessiver Zustand, was bedeutet, dass beide Elternteile den Defekt tragen müssen Gen und es besteht eine Wahrscheinlichkeit von eins zu vier, dass ein Kind betroffen ist (MIM 249100). Es gibt eine weniger häufige autosomal dominant Formular (MIM 134610).
Molekular Biologie und Genetik
Familiäres Mittelmeerfieber ist in der Regel darauf zurückzuführen Mutationen beim MittelmeerfieberMEFV) oder Pyrin-Gen befindet sich in Chromosom 16 (16p13.3). Mutationen in der MEFV Gen wurde im 80% der typischen Fälle gefunden. Mehr als 100 verschiedene krankheitsbedingte Mutationen wurden identifiziert. Am häufigsten berichtet Mutation, M694V, ist mit schwerer Krankheit verbunden und Entwicklung der sekundären Amyloidose. Andere häufige Mutationen sind M680I und V726A. 20% der aschkenasischen Juden tragen das E148Q MEFV Mutation.
das MEFV genetische Codes für a Protein genannt Pyrin (auch bekannt als Marenostrin), ausgedrückt hauptsächlich durch entzündlich weiße Blutkörperchen als Neutrophile und Eosinophile. Die normale Form von Pyrin unterdrückt Entzündungen, indem es entzündungsfördernde Medikamente herunterreguliert. Zytokine (zellulärer Bote Protein), Hochregulierung von Antiphlogistikum Zytokine und verhindern die Produktion von Interleukin 1beta (IL-1β) durch Inflammasomen. Der Mangel an funktionellem Pyrin löst die Entzündungskaskade aus.
Es gibt auch Umweltauswirkungen auf die Demonstrationen des familiären Mittelmeerfiebers. Beispielsweise haben Armenier mit familiärem Mittelmeerfieber, die in Armenien leben, a Vorfall Amyloidose von 25 auf 48%, während in den USA lebende Armenier eine Amyloidose-Inzidenz von 0% haben.
Was sind die klinischen Merkmale des familiären Mittelmeerfiebers?
Die häufigsten klinischen Merkmale des familiären Mittelmeerfiebers sind:
- Fieberkrämpfe, die 1 bis 3 Tage dauern
- starke Bauch-, Brust- und/oder Gelenkschmerzen
- Erysipel-ähnliche Veränderungen an den Unterschenkeln.
Der Beginn liegt fast immer vor dem 30. Kinder unter 2 Jahren haben oft nur Fieber und Fortschritt zu typischeren Attacken im Alter von 5 Jahren.
Die Eigenschaften des Wiederkehrenden akut Familiäre Mittelmeerfieber-Episoden sind in der folgenden Tabelle beschrieben.
| Symptom | Eigenschaften |
|---|---|
| Fieber |
|
| Bauchschmerzen |
|
| Brustschmerzen |
|
| Gelenkschmerzen und -schwellungen (Arthralgie, Arthritis) |
|
| Haut Eruptionen |
|
| Hodenschmerzen (Orchitis) |
|
| Vergrößerte Milz (Splenomegalie) |
|
| Muskelschmerzen (Myalgie) |
|
| Neurologisch Symptome |
|
| Amyloidose |
|
Was sind die Merkmale des Hautausschlags?
| Ähnlich wie Erysipel Verletzungen |
|
| Henoch-Schönlein lila |
|
| Unspezifische Purpura |
|
| Polyarteritis nodosa |
|
| Angioödem |
|
| Rötung der Handflächen |
|
| Raynauds Phänomen |
|
Was löst die Angriffe aus?
Die Häufigkeit der Episoden ist variabel, von einer Woche bis zu mehreren Jahren. Angriffe können ausgelöst werden durch:
- Übung
- Infektion
- Menstruation
- Betonen.
Akute Attacken lösen sich spontan auf. Zwischen den Episoden ist die Gesundheit normal.
Wie wird das familiäre Mittelmeerfieber diagnostiziert?
Die Diagnose des familiären Mittelmeerfiebers erfolgt anhand der Tel-Hahomer-Kriterien:
- Die Diagnose ist definitiv, wenn 2 Hauptkriterien oder 1 Hauptkriterium + 2 Nebenkriterien erfüllt sind.
- Die Diagnose ist wahrscheinlich, wenn 1 Haupt- + 1 Nebenkriterium erfüllt sind.
Hauptkriterien
- Wiederkehrende Fieberschübe in Verbindung mit Peritonitis, Pleuritis oder Synovitis
- Typ-AA-Amyloidose ohne prädisponierende Erkrankung
- Günstige Reaktion auf tägliches Colchicin.
Kleinere Kriterien
- Wiederkehrende Fieberschübe
- Ähnlich wie Erysipel Erythem
- Positive Anamnese von familiärem Mittelmeerfieber bei einem Verwandten ersten Grades.
Untersuchungen beim familiären Mittelmeerfieber
Bluttests während eines Angriffs können zeigen:
- Erhöhte Anzahl weißer Blutkörperchen (Leukozytose), Erythrozyten Sedimentationsrate (ESR), Serum Fibrinogen, C-reaktives Protein (CRP)
- Erhöhtes Serum-IgD in einem 10%.
Röntgenaufnahmen des Abdomens zeigen oft mehrere Flüssigkeitsspiegel, die auf ein „akutes Abdomen“ hindeuten
Eine Haut Biopsie von Erysipel Verletzung kann ein schweres zeigen infiltrieren Neutrophile (weiße Blutkörperchen). Leukozytoklast Vaskulitis es ist in gesehen Biopsien Henoch-Schönlein-Purpura oder Polyarteritis nodosa-ähnliche Läsionen.
Untersuchungen auf Myalgie (Muskelschmerzen) zeigen normale Muskeln Enzyme, Elektromyographie (EMG) und Muskelbiopsie.
Amyloidose zeigt sich am häufigsten als Proteinurie ohne rote Blutkörperchen oder erhöhten Blutdruck.
Die Gensequenzierung kann von spezialisierten Labors als ortsgerichtete Mutationsanalyse (Suche nach den häufigsten Mutationen) oder als Sequenzanalyse ausgewählter Exons (Suche nach Exon 10 und möglicherweise anderen) durchgeführt werden. Sobald die genetischen Mutationen beim Patienten identifiziert wurden, wird der Trägerstatus der Eltern, das Screening von anderen Blutsverwandten ersten Grades, selbst wenn asymptomatischund vorgeburtlich Diagnose festgestellt werden kann. Die Identifizierung asymptomatischer, aber genetisch betroffener Personen bedeutet, dass eine Behandlung eingeleitet werden kann, um die Entwicklung einer Amyloidose zu verhindern.
Wie wird das familiäre Mittelmeerfieber behandelt?
Colchicin, lebenslang täglich oral eingenommen, ist das Mittel der Wahl bei familiärem Mittelmeerfieber bei:
- reduzieren Sie die Häufigkeit von Angriffen
- die Schwere der Attacken reduzieren
- sekundär verhindern systemisch Amyloidose.
Colchicin ist potenziell giftig Aus diesem Grund ist es sehr wichtig, keine übermäßige Dosis einzunehmen.
Die Colchicin-Dosis wird schrittweise auf eine Höchstdosis von 2,5 mg/Tag gesteigert. Die Dosis wird normalerweise durch die Häufigkeit und Schwere der Attacken bestimmt. Colchicin führt zu einer deutlichen Verbesserung der Symptome bei den Patienten 90-95% und 75% zeigt a Remission. Es ist nicht bekannt, wie es funktioniert.
andere Behandlung
Akute Attacken werden mit behandelt Nicht-steroidale entzündungshemmende Medikamente (NSAIDs) und Schmerzlinderung.
Längeres Fieber mit Muskelschmerzen reagiert auf systemische Kortikosteroide.
Der häufigste Grund für das Nichtansprechen auf Colchicin ist eine schlechte Compliance aufgrund von Magen-Darm-Beschwerden. 5-10% reagiert jedoch nicht auf Colchicin und dies kann darauf zurückzuführen sein ABCB1 Gen Polymorphismen das die Aufnahme von Colchicin durch mononukleäre Zellen beeinflusst.
Thalidomid und biologische Wirkstoffe, wie täglich subkutan Anakinra (ein Interleukin-1 Empfänger -Antagonist) oder Etanercept, können zur Vorbeugung von Attacken und der Entwicklung einer Amyloidose in Betracht gezogen werden. Um die Schwere eines Anfalls zu verringern, wurde berichtet, dass eine Einzeldosis Methylprednisolon oder Anakinra zu Beginn der Episoden die Symptome lindert.