Was ist Angioimmunoblasten? T-Zelle Lymphom?
Angioimmunoblastisches T-Zell-Lymphom ist selten Knoten peripher T-Zell-Lymphom (PTCL), das produziert systemisch Symptome und schwerwiegende Immunschwäche [1]. Manchmal verursacht es eine Eruption.
Angioimmunoblastisches T-Zell-Lymphom ist charakterisiert histologisch zum polymorph infiltrieren hauptsächlich mit Lymphe Knoten, ein Proliferation Hoch endothelial Venolenund follikulär dendritische Zellen [2].
Wer bekommt ein angioimmunoblastisches T-Zell-Lymphom?
Angioimmunoblastisches T-Zell-Lymphom ist selten.
- Periphere T-Zelle Lymphome umfassen 10 bis 15% von allen lymphoid maligne Erkrankungen [1,3]
- Angioimmunoblastisches T-Zell-Lymphom macht 15 bis 20% peripherer T-Zell-Lymphome und 1 bis 2% Non-Hodgkin-Lymphome aus [2,4].
Angioimmunoblastisches T-Zell-Lymphom betrifft im Allgemeinen ältere Erwachsene [4]. das Median Das Diagnosealter beträgt 62 bis 65 Jahre. Vorfall es ist das gleiche bei Männern und Frauen [4].
Es wurden keine Risikofaktoren für ein angioimmunoblastisches T-Zell-Lymphom identifiziert.
Was verursacht angioimmunoblastisches T-Zell-Lymphom?
Die Ursprungszelle des angioimmunoblastischen T-Zell-Lymphoms ist die follikuläre Helfer-T-Zelle. [4]. Der Böse Transformation wurde mit in Verbindung gebracht Mutationen beim epigenetisch RegulierungsbehördenTET2, IDH2 und DNMT3A), das Homolog von Ras Gen Familienmitglied A (RHOA) und T-Zellen Empfänger Route (CD28, FYN, PLCG1, CARD11, P13K Elemente, CTNNB1und GTF2I) [4].
Angioimmunoblastisches T-Zell-Lymphom wird durch Überexpression von gefördert angiogen Mediatoren (Faktoren, die regulieren Entwicklung von Blutgefäße), wie vaskulär endothelial Wachstumsfaktor (VEGF) und Interleukin (IL) -8 und Zytokine Was sind Sie entzündungshemmend (IL6, IL18), Immunsuppressivum (IL10) oder Proliferation induzieren (IL21) [4,5].
Infektionskrankheiten, die mit angioimmunoblastischem T-Zell-Lymphom assoziiert sind, umfassen das Epstein-Barr-Virus (EBV, die Ursache der infektiösen Mononukleose), das humane Herpesvirus 6 (HHV-6) (die Ursache der Roseola), HHV-8 (die Ursache von Kaposi Sarkom), menschlicher Immunschwächevirus (HIV), bakteriell Infektionen und Pilzinfektionen [4].
Die Rolle des Epstein-Barr-Virus
EBV setzt auf Leben hartnäckig Infektion im B-Zellen. Eine durch angioimmunoblastisches T-Zell-Lymphom verursachte Immunschwäche reaktiviert EBV [4,6–8].
- EBV-infizierte B-Zellen sind bei 58 bis 97% von Patienten mit angioimmunoblastischem T-Zell-Lymphom vorhanden [8,9].
- EBV (und möglicherweise HHV-6) kann eine Rolle bei der Förderung des Fortschreitens der Krankheit durch Modulation von Zytokinen spielen. Chemokineund Membranrezeptoren [6].
- Es wird angenommen, dass EBV die Lebensdauer von B-Zellen verlängert und verursachen kann genetisch Aberrationen wie die Prävention von induziertem c-myc Apoptose, was zu einem B-Zell-Lymphom führt [4,8,10]. Siehe im Zusammenhang mit dem Epstein-Barr-Virus lymphoproliferativ Störungen.
Was sind die klinischen Merkmale des angioimmunoblastischen T-Zell-Lymphoms?
Mehr als 70% von Patienten mit angioimmunoblastischem T-Zell-Lymphom haben konstitutionelle Symptome. Dazu gehören Fieber, Schüttelfrost, Nachtschweiß, leichte Schmerzen, Gewichtsverlust, Arthralgienund in 50% ein Ausschlag [11].
Lymphadenopathie und Hepatosplenomegalie Sie sind normalerweise bei der Prüfung anwesend. Pleural Ergüsse, Aszites, neurologisch Gastrointestinale Anzeichen und Symptome sind seltener [4].
Haut Merkmale des angioimmunoblastischen T-Zell-Lymphoms
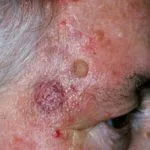
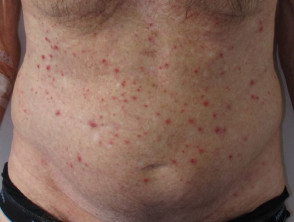
Angioimmunoblastisches T-Zell-Lymphom weist am häufigsten einen unspezifischen Ausschlag auf [11]. Es kann auch dazu führen Papeln, Knötchen, Platten, Geschwüre, Petechienund selten Erythrodermie [8,11,12].
Hautausschlag im Zusammenhang mit angioimmunoblastischem T-Zell-Lymphom
Der Patient hatte ein EBV-assoziiertes T-Zell-Lymphom
Was sind die Komplikationen eines angioimmunoblastischen T-Zell-Lymphoms?
Patienten mit angioimmunoblastischem T-Zell-Lymphom haben insbesondere ein erhöhtes Risiko für sekundäres B-Zell-Lymphom diffus großes B-Zell-Lymphom (DLBCL) und seltener Hodgkin-Lymphom oder Plasmozytom [13].
Es gibt 30 dokumentierte Fälle von EBV-assoziierten B-Zell-Lymphomen bei Patienten mit angioimmunoblastischem T-Zell-Lymphom (November 2019) [14–16].
Diffuses EBV-induziertes großzelliges B-Zell-Lymphom kann auch Hautzeichen aufweisen, wie z. B. ulzerierte Papeln, Knötchen und Abszesse [16].
Wie wird ein angioimmunoblastisches T-Zell-Lymphom diagnostiziert?
Labor Anomalien einschließen:
- Anämie (häufig hämolytischmit einem positiven Coombs-Test)
- Eosinophilie
- Polyklonale Hypergammaglobulinämie
- Antinukleär Antikörper (ANA)
- Kalt Agglutinine
- Kryoglobulinämie
- Zirkulierende Immunkomplexe
- Hoch Serum Lactatdehydrogenase (LDH) und Beta-2-Mikroglobulin
- Erhöhtes EBV.
Peripheres Blut Leukozytose mit Lymphozytose ist ungewöhnlich. Durchflusszytometrie kann zum Nachweis verwendet werden aberrant T-Zellen [17].
Die Diagnose eines angioimmunoblastischen T-Zell-Lymphoms wird üblicherweise von gestellt Lymphknoten Biopsie.
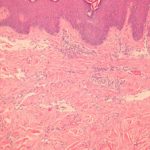
Es gibt fünf angioimmunoblastische T-Zell-Lymphome histopathologisch Muster auf der Haut.
- Eine flache perivaskulär Infiltrat bestehend aus Eosinophile und Lymphozyten dieser Mangel Atypie (das häufigste Muster)
- Ein spärliches perivaskuläres Infiltrat mit atypisch Lymphozyten
- Ein dichtes oberflächliches und tiefes Infiltrat von pleomorph Lymphozyten
- Vaskulitismit oder ohne atypische Lymphozyten
- Nekrotisierend Granulome.
Histologie des angioimmunoblastischen T-Zell-Lymphoms
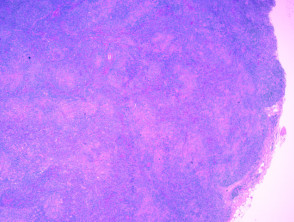
H & E x 20
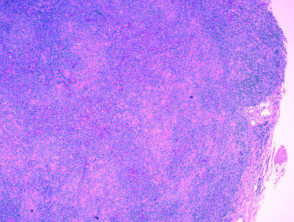
H & E x 40
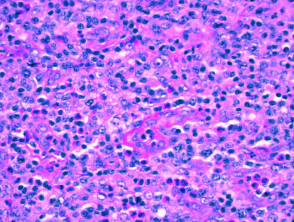
H & E x 400
Der Immunphänotyp von neoplastisch entspricht typischerweise follikulären T-Helferzellen (CD3 +, CD4 +, CD8−, CD10 +, PD-1 +, ICOS +, Bcl-6 +, Chemokin Ligand CXCL13 +) mehr Gruppen von CD21 + follikuläre dendritische Zellen.
Positives EBV gefunden in a Immunhistochemie Der Fleck ist im reiferen höher Verletzungen und bildet gelegentlich unscharfe Muster [18].
Immunhistochemie des angioimmunoblastischen T-Zell-Lymphoms
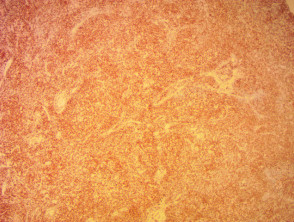
Anti-CD3 x 20
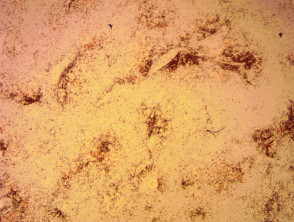
Anti-CD 20 x 20
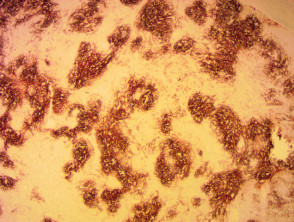
Anti CD 21 x 20
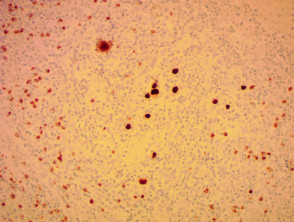
Anti-Epstein-Lauf x 200
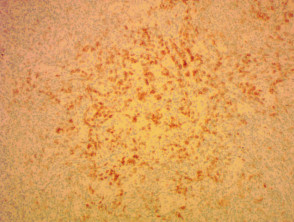
Anti-PD1 x 100
Welches ist das Differenzialdiagnose für kutanes angioimmunoblastisches T-Zell-Lymphom?
Die Differentialdiagnose des kutanen angioimmunoblastischen T-Zell-Lymphoms hängt von der Morphologie des Hautausschlags, kann aber umfassen:
- Arzneimittel Eruption
- Viral Exanthem
- Atypische mykobakterielle Infektion oder andere opportunistische Infektion
- Andere Ursachen für Erythrodermie.
Was ist die Behandlung für angioimmunoblastisches T-Zell-Lymphom?
Aufgrund der Seltenheit des angioimmunoblastischen T-Zell-Lymphoms wird die Behandlung im Allgemeinen von prospektiven Phase-II-Studien geleitet [4].
- Die häufigste Behandlung basiert auf Anthracyclin. Chemotherapie wie CHOP (Cyclophosphamid, Doxorubicin, Vincristin und Prednison / Prednisolon).
- Follow-up-Konsolidierung mit autologen Stammzellen Transplantation hat vielversprechende Ergebnisse gezeigt [19].
- Histon-Deacetylase (HDAC) -Tests Inhibitoren, Hypomethylierungsmittel und Anti-CD30-Antikörper sind noch nicht abgeschlossen [20–23].
Was ist das Ergebnis eines angioimmunoblastischen T-Zell-Lymphoms?
Angioimmunoblastisches T-Zell-Lymphom ist normalerweise aggressiv mit einem mittleren Überleben von weniger als 3 Jahren, selbst bei intensiver Behandlung. Patienten, bei denen normalerweise eine Erkrankung im späten Stadium III-IV vorliegt und deren Überlebensraten nach 5 Jahren als 33% und nach 7 Jahren als 29% angegeben wurden [18].
- Die Standard-CHOP-Chemotherapie hat eine Gesamtansprechrate von 70% bis 80% und ein progressionsfreies 5-Jahres-Überleben von 10% bis 20% [8,24].
- Follow-up-BEAM (Carmustin, Etoposid, Cytarabin, Melphalan) und autologe Stammzelltransplantation weisen eine progressionsfreie 5-Jahres-Überlebensrate von etwa 40% auf [25].
Zirkulierendes EBV DNA Pegel können gesteuert werden; höhere Werte sind mit ärmeren verbunden Prognose. Das Vorhandensein von EBV in Gangliontumoren ändert jedoch nichts an der Prognose.
Sekundäre B-Zell-Lymphome haben typischerweise eine schlechte Prognose. Acht von 30 Patienten mit EBV-assoziiertem B-Zell-Lymphom überlebten länger als 12 Monate [14–16].