Was ist Gardner? Syndrom?
Das Gardner-Syndrom ist eine Variante von 'Familie adenomatös Polyposis '(FAP), eine Erbkrankheit, die durch gastrointestinale Polypen, multiple Osteome (gutartig Knochentumoren) und Haut und Weicher Stoff Tumoren Polypen neigen dazu, sich in der Pubertät mit einem durchschnittlichen Diagnosealter von etwa 25 Jahren zu bilden. Bei fast allen Patienten Polypen Fortschritt zu Malignität, was zu kolorektalen Krebs Eine rechtzeitige Erkennung ist daher unerlässlich.
Was verursacht das Gardner-Syndrom?
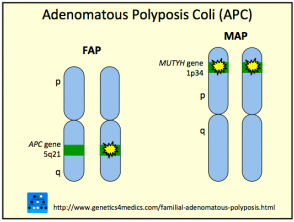
Das Gardner-Syndrom ist auf zurückzuführen Mutationen über ihn APC Gen im Chromosom 5q22. Das Gen spielt eine Rolle in Tumor Unterdrückung. Das Gardner-Syndrom wird als vererbt autosomal dominante Eigenschaft für eine betroffene Person, eine 50%-Chance zu haben, das Gen auf jedes ihrer Kinder zu übertragen.
Genetik des Gardner-Syndroms *
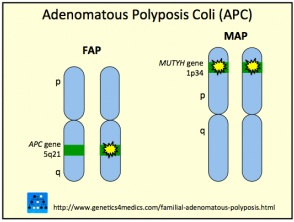
Gardner-Syndrom
* Mit freundlicher Genehmigung von Genetics 4 Medics
Was sind die klinischen Merkmale des Gardner-Syndroms?
Die klinischen Merkmale des Gardner-Syndroms können in zwei Typen unterteilt werden: Haut- und nicht kutan. Das bemerkenswerteste kutane Merkmal des Gardner-Syndroms ist das Auftreten von Epidermoidzysten. Diese Zysten können sein differenziert von gewöhnlichen Epidermoidzysten durch die folgenden Faktoren:
- Epidermoid Aufnahme Gardner-Syndrom-Zysten (50-65%) treten in einem früheren Alter (um die Pubertät) auf als gewöhnliche Zysten.
- Epidermoidzysten treten an weniger häufigen Stellen wie Gesicht, Kopfhaut und Extremitäten auf als gewöhnliche Zysten.
- Zysten sind bei mehr als der Hälfte der Patienten mit Gardner-Syndrom in der Regel mehrfach.
- Wie bei gewöhnlichen Epidermoidzysten sind es normalerweise die Zysten beim Gardner-Syndrom asymptomatisch (keine Symptome), jedoch in einigen Fällen können sie sein juckend (juckend) und / oder entzündet und kann brechen.
- Manchmal haben Zysten hybride Merkmale mit Pilomatricoma Histopathologie
Andere Hautmerkmale umfassen Desmoidzysten, Myome, Lipome, Leiomyome, Neurofibrome und pigmentiert Haut Verletzungen.
Zu den nicht kutanen Merkmalen gehören:
- Gastrointestinale Polypen, die sich fast immer zu Dickdarmadenokarzinomen (Dickdarmkrebs) entwickeln.
- Osteome: Diese gutartigen Knochentumoren sind für die Diagnose des Gardner-Syndroms unerlässlich. Sie kommen am häufigsten in der Unterkiefer (Kiefer) kann aber auch in den Schädel und die langen Knochen hineinwachsen.
- Dental Anomalien - Zusätzlich zu Osteomen im Kiefer können andere Zahnanomalien auftreten, z. B. nicht durchgebrochene zusätzliche Zähne und Hohlräume
- Multifokale pigmentierte Läsionen der Fundus im Auge - gesehen im 80% von Patienten. Diese Läsionen können kurz nach der Geburt vorliegen und der erste Marker für die Krankheit sein.
Wie wird das Gardner-Syndrom diagnostiziert?
Das Gardner-Syndrom wird anhand der folgenden Merkmale diagnostiziert.
- ≥ 100 kolorektale Polypen ODER ≤ 100 Polypen und ein Familienmitglied mit familiärer adenomatöser Polyposis oder Gardner-Syndrom
- Osteome
- Weicher Stoff Tumoren wie Epidermoidzysten, Myome und Desmoide Tumoren
- APC Mutation.
Radiologic Studien sind für Patienten und Angehörige mit Verdacht auf Gardner-Syndrom unerlässlich.
- Bilder des langen Knochens können Osteome zeigen.
- Die Darstellung des Kiefers in jungen Jahren kann subtile Defekte aufweisen.
- Augenuntersuchungen in einem frühen Alter können pigmentierte Läsionen im Fundus erkennen.
- Darmspiegelung und ein anderer angreifend Tests zur Überprüfung auf Polypen alle ein bis zwei Jahre.
Was ist die Behandlung für das Gardner-Syndrom?
Die Behandlung von Epidermoidzysten beim Gardner-Syndrom ähnelt der bei normalen Zysten verwendeten und beinhaltet Exzision. Gelegentlich können intraläsionale Steroidinjektionen verwendet werden, wenn die Zysten entzündet sind.
Die chirurgische Entfernung von Magen-Darm-Polypen wird aufgrund des hohen Polypenrisikos empfohlen Entwicklung bei Krebs. Patienten benötigen regelmäßige Koloskopien.
Osteome können auch nur dann entfernt werden, wenn sie stark deformiert sind oder ein Ärgernis darstellen.