Was ist Ehlers - Danlos? Syndrom?
Das Ehlers-Danlos-Syndrom (EDS) ist eine Gruppe von Erbkrankheiten, an denen a genetisch Defekt in Kollagen oder Bindegewebe Synthese und Struktur [1]. Das führt zu:
- Zerbrechliche und hyperelastische Haut
- Instabile und überstreckbare Gelenke (hypermobil)
- Zerbrechliches Gewebe und Blutgefäße.
Ehlers-Danlos-Syndrom
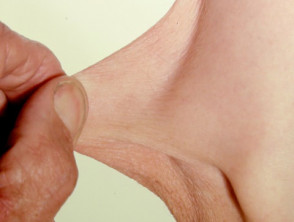
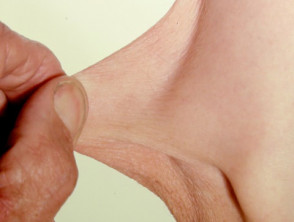
Hyperelastizität der Ellenbogenhaut
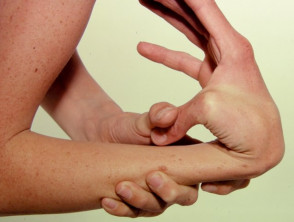
Hypermobilität
Prellungen und Narben
Wer bekommt das Ehlers-Danlos-Syndrom?
Die Gesamthäufigkeit von EDS beträgt ungefähr 1 zu 5000. EDS kann bei Männern und Frauen aller Rassen auftreten und manifestiert sich normalerweise zuerst im Leben von Kindern und jungen Erwachsenen.
EDS hat autosomal dominant und autosomal rezessiv Vererbungsmuster.
- das autosomal dominant Die Krankheit tritt auf, wenn nur eine Kopie von a Gen es ist erforderlich, eine Bedingung zu verursachen. Es besteht eine 50%-Chance, dass ein Nachwuchs das abnormale Gen von einem betroffenen Elternteil erbt.
- Eine autosomal-rezessive Erkrankung erfordert zwei Kopien eines abnormalen Gens, um eine Erkrankung zu verursachen. daher ein Kind von zwei Jahren Träger Eltern sind einem Risiko für 25% und 50% von ausgesetzt Entwicklung Krankheit bzw. Träger einer Krankheit sein.
Subtypen des Ehlers-Danlos-Syndroms
Gemäß der Internationalen Klassifikation für Ehlers-Danlos-Syndrome 2017 gibt es dreizehn EDS-Subtypen, die nach klinischen Merkmalen, Vererbungsmuster und klassifiziert sind molekular genetische Defekte.
Jeder von ihnen ist eine andere Störung, die in einer Familie "wahr" ist. Dies bedeutet, dass Mitglieder einer von EDS betroffenen Familie dieselben Merkmale aufweisen. Einige Fälle passen nicht perfekt zu einem bekannten EDS-Typ, und ein Patient kann Merkmale von mehr als einem Typ aufweisen. Klinisch Demonstrationen und sein Schweregrad kann selbst innerhalb derselben Familie erheblich variieren.
Hypermobiles EDS ist der häufigste Subtyp von EDS (MIM 130020), gefolgt von klassischem EDS (MIM 130000) und vaskulär EDS (MIM 130050). Die anderen Arten von EDS sind sehr selten.
Siehe Subtypen des Ehlers-Danlos-Syndroms [Tabelle in einer neuen Registerkarte anzeigen] [2,3]
Was verursacht das Ehlers-Danlos-Syndrom?
Kollagen ist einer der Hauptbausteine des Körpers. Ist ein Protein Es ist weit verbreitet in der Struktur vieler Gewebe und Organe im Körper. Es gibt verschiedene Arten von Kollagen mit jeweils unterschiedlichen Eigenschaften. Kollagen kann Kraft und festen Halt bieten, es kann dehnbar sein, um Bewegung zu ermöglichen, oder es kann verwendet werden, um Dinge zusammenzuhalten.
Ein genetischer Defekt kann zu reduzierten Kollagenmengen, zu einer Störung des Kollagens (Kollagen wird normalerweise in Bündeln angeordnet) und zu Veränderungen der Größe und Form des Kollagens führen. Der Subtyp von EDS hängt davon ab, wie Kollagen Stoffwechsel wurde betroffen. Beispielsweise wird vaskuläres EDS durch eine verminderte oder fehlende Kollagen-Typ-III-Synthese verursacht.
Es gibt einige Arten von EDS, die das Ergebnis anderer sind die extrazelluläre Matrix Störungen und ihre Bestandteile (wie Glykosaminoglykane) und Defekte in intrazellulär Wird bearbeitet.
Was sind die klinischen Merkmale des Ehlers-Danlos-Syndroms?
Anzeichen und Symptome unterscheiden sich in Art und Schweregrad zwischen den verschiedenen Arten von EDS. Nachfolgend finden Sie eine Zusammenfassung der klinischen Merkmale von systembasiertem EDS.
Gesichtszüge
- Eine dünne und enge Nase
- Prominente Augen
- Ohren ohne Lappen
Parodontale Beteiligung
- Schwer und hartnäckig Entzündung Gummi
- Mangel an klebrigem Gummi
Bewegungsapparat System
- Hypermobilität: Die Gelenke biegen sich mehr als gewöhnlich. Die Finger sind am stärksten betroffen, aber alle Gelenke können betroffen sein. Angeboren oder wiederkehrend Gelenkversetzungen oder Subluxationen können insbesondere in den Schultern, Hüften und Knien auftreten.
- Talipes angeborener Equinovarus (Klumpfuß) - a Entwicklung Deformität des Fußes, wo ein oder beide Füße nach innen und unten gedreht sind.
- Angeborene oder früh einsetzende Kyphoskoliose: eine abnormale Krümmung der Wirbelsäule.
- Arachnodaktylie - ungewöhnlich lange und dünne Finger und Zehen
- Angeboren oder mild, spät einsetzend Hypotonie und Bruttomotorverzögerung
Hypermobilität beim Ehlers-Danlos-Syndrom
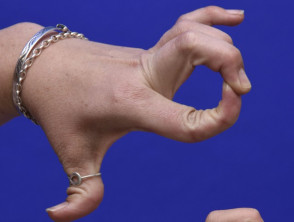
Hypermobile Gelenke
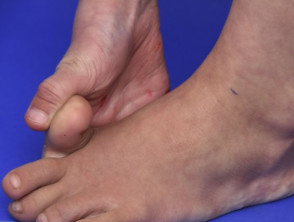
Hypermobile Gelenke
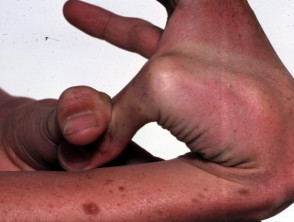
Hypermobile Gelenke
Haut
- Überstreckbarkeit der Haut: Es ist leicht, die Haut vom Körper zu trennen, und nach ihrer Freisetzung zieht sie sich in ihren ursprünglichen Zustand zurück.
- Schlank und durchscheinend Haut mit sichtbaren darunter liegenden Blutgefäßen.
- Haut Zerbrechlichkeit - Die Haut bricht und reißt leicht.
- Prellungen und Prellungen kann nach geringfügigen Verletzungen auftreten und kann an Stellen gesehen werden, die nicht dazu neigen Trauma.
- Vorzeitig gealtertes Erscheinungsbild dünner und faltiger Haut, insbesondere an Händen und Füßen.
- Atrophisch Haut- Narben - Wunden heilen sehr langsam, was zu Narben im Fischmaul oder im Zigarettenpapier führt.
- Epicanthic Falten Lassen Sie den Nasenrücken breit erscheinen.
- Mollusken-Pseudotumoren: Dies sind kleine schwammige Wucherungen mit einem Durchmesser von 2 bis 3 cm an Druckpunkten wie Knien und Ellbogen.
- Subkutan Sphäroide: verkalkte Fettlappen, die auf eine unzureichende Blutversorgung zurückzuführen sind.
- Knötchen kleine, feste Klumpen direkt unter der Hautoberfläche, die am häufigsten an Armen und Schienbeinen auftreten.
- Pretibial Platten.
Überstreckbarkeit beim Ehlers-Danlos-Syndrom
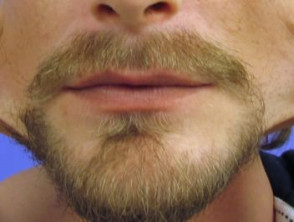
Hyperelastizität der Wangenhaut.
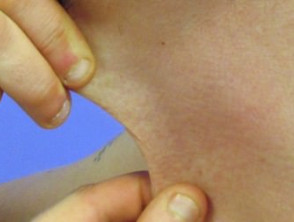
Hyperelastizität der Halshaut.
Hyperelastizität der Ellenbogenhaut
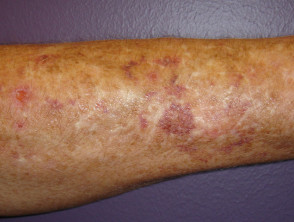
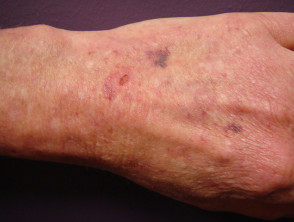
Prellungen beim Ehlers-Danlos-Syndrom
Ehlers-Danlos-Syndrom
Ehlers-Danlos-Syndrom
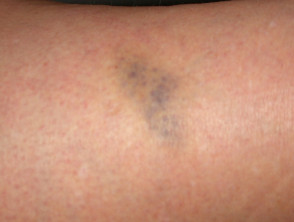
Prellungen
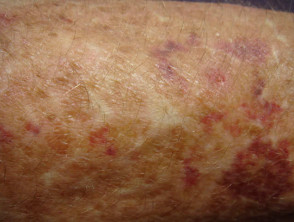
Atrophische Narben beim Ehlers-Danlos-Syndrom
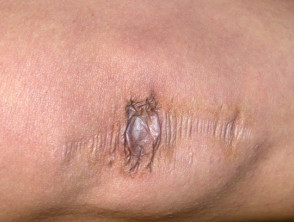
Umfangreiche Narbenbildung bei einem Patienten mit Ehlers-Danlos-Syndrom.
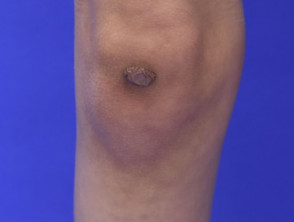
Atrophische Narbe
Hypopigmentierte Narben
Haut-Ehlers-Danlos-Syndrom
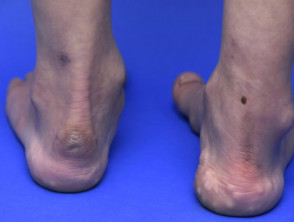
Pseudotumor im Knöchel
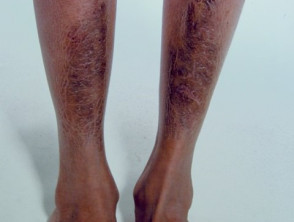
Pretibialplatten
Weitere Bilder des Ehlers-Danlos-Syndroms.
Auge
- dünn Hornhautmit oder ohne Pause
- Blau sklerotisch
- Keratokonus: Die Hornhaut ist kegelförmig
- Keratoglobus - der kugelförmig Ausbeulung und Ausdünnung der Hornhaut
Interne Kollagendefekte
- Herzgeräusch (Mitralklappenprolaps) und geschwächte Darmwände. Arterienund Gebärmutter, die gebrochen werden kann
- Autonom Funktionsstörung - vorübergehend Bewusstlosigkeit, Herzklopfen, Schwindel, Brustschmerzen und Atemnot
- Haltungsorthostatik Tachykardie Syndrom (POTS): schneller Herzschlag, der insbesondere beim Ändern der Körperhaltung oder im Stehen auftritt
- Wiederkehrend Pneumothorax - Dies kann die erste Präsentation von EDS sein.
Was sind die Komplikationen des Ehlers-Danlos-Syndroms?
Mögliche Komplikationen des Ehlers-Danlos-Syndroms sind:
- Chronisch Gelenkschmerzen
- Schlechte Wundheilung
- Versagen des chirurgischen Wundverschlusses
- Früher Start Arteriitis
- Vorzeitiger Bruch der fetalen Membran
- Gefäß- und Hohlorganruptur
- Ballonruptur, Netzhautablösung und Glaukom
- Epilepsie [4].
Wie wird das Ehlers-Danlos-Syndrom diagnostiziert?
Der Verdacht auf das Ehlers-Danlos-Syndrom sollte zu einer sorgfältigen Anamnese und Untersuchung führen. Für jeden der 13 EDS-Subtypen wurden Haupt- und Nebenkriterien entwickelt [3]. Die endgültige Diagnose einer bestimmten Variante erfordert Gewebe Biopsie und molekulargenetische Analyse aufgrund der Vielzahl molekularer Defekte und der klinischen Überlappung zwischen EDS-Subtypen.
Krankengeschichte
Detaillierte persönliche und familiäre Geschichte der Muskeln, Okular, Haut-, Zahn-, Herz- und Atemwegserkrankungen.
Klinische Untersuchung
- Umfassende Hautuntersuchung: Beurteilung der Überstreckbarkeit der Haut und abnormaler Narben.
- Der Beighton Rahmen - Beurteilung der Hypermobilität eines Gelenks, bei der eine Punktzahl von 5 oder mehr anzeigt weit verbreitet Gelenkhypermobilität.
Forschung
- Hautbiopsie für Histologie, Fibroblasten Kulturund Elektron Mikroskopie
- Bei Verdacht auf kyphoskoliotisches EDS wird eine Urinprobe entnommen, um das Verhältnis von Lysylpyridinolin zu Hydroxylysylpyridinolin-Vernetzungen zu messen.
Molekulargenetische Tests bestätigen die Diagnose, außer im Fall von hypermobilem EDS, das eine unbekannte genetische Basis hat und daher eine klinische Diagnose ist, die alle klinischen Kriterien erfüllen muss.
Welches ist das Differenzialdiagnose für Ehlers-Danlos-Syndrom?
Zu den Bedingungen, die mit EDS verwechselt werden können, gehören:
- Unvollkommene Osteogenese
- Loeys-Dietz-Syndrom
- Skelettdysplasien
- Mukopolysaccharidose
- Haut Laxa-Syndrom
- Elastisches Pseudoxanthom
- Angeborene muskulöse Ulrich Dystrophie
- Bethlem Myopathie
- Larsen-Syndrom
- Achondroplasie
- RIN2-Syndrom
- Marfan-Syndrom
- Chondrodysplasie
- Angeborene Myopathien
- Freeman-Sheldon-Syndrom.
Was ist die Behandlung für das Ehlers-Danlos-Syndrom?
Verwaltungsrichtlinien hängen vom EDS-Subtyp ab.
- Ergreifen Sie vorbeugende Maßnahmen gegen schwerwiegende und lebensbedrohliche Komplikationen.
- Patienten sollten in Betracht ziehen, ein MedicAlert-Armband zu tragen, um ihren EDS-Status mitzuteilen.
- Alle Patienten mit EDS sollten zur genetischen Beratung und zu geeigneten Spezialisten überwiesen werden.
Allgemeine Maßnahmen
- Kinder können von Schutzpolstern und Bandagen profitieren.
- Wenden Sie sich an Sportarten oder Aktivitäten, die dies betreffen intrakraniell Druck (z. B. Trompetenspiel) sollte vermieden werden, um ein übermäßiges Trauma zu vermeiden.
- Der Patient kann von Übungen mit geringem Widerstand und Physiotherapie profitieren.
- Erwägen Sie Kalzium- und Vitamin D-Präparate, um die Knochendichte zu optimieren.
- Bieten Sie nach der Diagnose psychologische Unterstützung an.
- Die Patienten müssen ein medizinisches Warnarmband mit der Aufschrift "EDS-Typ" tragen.
Medizin
- Ascorbinsäure (Vitamin C) wurde verwendet, um Blutergüsse zu reduzieren.
Chirurgie und Wundversorgung
- Die Operation sollte angesichts des Risikos von Gefäßkomplikationen mit Vorsicht durchgeführt werden.
- Wunden sollten vorsichtig mit spannungsfreien Nähten verschlossen werden.
- Tiefe Stiche werden empfohlen; Lassen Sie flache Nähte für längere Zeit, um die Heilung zu ermöglichen.
Die Schwangerschaft
- Eine geburtshilfliche Überwachung wird während der gesamten Schwangerschaft empfohlen, da das Risiko von Blutungen und Gefäßerkrankungen sowie chirurgischen und chirurgischen Störungen besteht Narkose Komplikationen.
- Frauen mit EDS haben ein erhöhtes Risiko für Frühgeburten nach der Geburt Blutung, Blasen- und Uterusprolaps, Bauchhernien und Wunddehiszenz.
- Angesichts des breiten Spektrums möglicher Auswirkungen und des Schweregrads jedes Subtyps gibt es keine Richtlinien für das geburtshilfliche Management von EDS. Daher werden Patienten von Fall zu Fall behandelt.
Überprüfung und Überwachung
- Der Patient sollte eine regelmäßige Blutdrucküberwachung haben und arteriell Krankheitsscreening zur Verringerung des Risikos von Blutungen und Gefäßkomplikationen.
- Nein-angreifend auf den Bildschirm bringen - Ultraschall, Computertomographie (Connecticut) scannen, Echokardiogrammund Magnetresonanztomographie (Magnetresonanz) kann durchgeführt werden, um mögliche Komplikationen zu identifizieren.
- Invasive Screening-Verfahren müssen sorgfältig abgewogen werden.
- Die Knochendichtescans der Dual-Energy-Röntgenabsorptiometrie (DEXA) sollten alle paar Jahre durchgeführt werden.
- KEDS-Patienten sollten regelmäßig einer Augenuntersuchung unterzogen werden, da bei ihnen das Risiko von Augenkomplikationen besteht.
Was ist das Ergebnis für Patienten mit Ehlers-Danlos-Syndrom?
das Prognose der Patienten mit Ehlers-Danlos-Syndrom variiert sogar innerhalb desselben Subtyps stark. Die meisten Patienten haben eine normale Lebenserwartung und kognitive Funktion. Menschen mit vaskulärem EDS haben möglicherweise eine kürzere Lebenserwartung und ein erhöhtes Risiko für einen plötzlichen Tod aufgrund des Bruchs eines inneren Organs und eines Hauptgefäßes.