Cos'è Sturge - Weber sindrome?
La sindrome di Sturge-Weber è rara, congenitae non ereditato neurocutaneo disturbo caratterizzato da capillare malformazione sulla pelle del viso (macchia di vino porto) e sui capillarivenoso malformazioni nel cervello e negli occhi [1].
Sindrome di Sturge-Weber
Sindrome di Sturge-Weber
Chi ha la sindrome di Sturge-Weber?
Si stima che circa un bambino su 20.000-50.000 nasca con la sindrome di Sturge-Weber [2]. Poiché è causato da a somatico mosaico mutazione, non è ereditato dai genitori.
Quali sono le cause della sindrome di Sturge-Weber?
La sindrome di Sturge-Weber è causata da una mutazione a mosaico somatico del GNAQ gene in cromosoma 9 [3]. Ciò significa che la mutazione nel gene si è verificata nelle cellule del corpo dopo la formazione dello zigote. GNAQ regola intracellulare percorsi di segnalazione. La mutazione provoca la formazione o la maturazione incontrollata di capillari nelle cellule colpite.
Quali sono le caratteristiche cliniche della sindrome di Sturge-Weber?
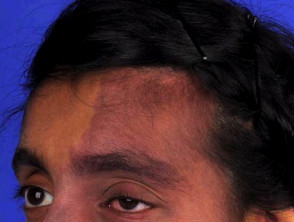
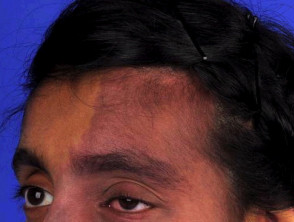
La sindrome di Sturge-Weber è caratterizzata da vascolare malformazioni al viso, agli occhi e al cervello delle persone colpite. Questi sono presenti alla nascita.
- Le macchie di vino porto sono il tipo più comune di malformazione vascolare, che colpisce circa tre bambini su 1.000, ma la maggior parte non è associata alla sindrome di Sturge-Weber. [4].
- Le macchie di vino porto nella sindrome di Sturge-Weber si trovano tipicamente nel distribuzione della prima e della seconda divisione del trigemino nervo sulla fronte e sulla palpebra superiore [5]. Possono anche interessare entrambi i lati del viso. [6].
- Malformazioni vascolari leptomeningee insorgono all'interno del cervello sullo stesso lato della macchia di vino porto.
- Le malformazioni vascolari leptomeningee possono anche verificarsi senza una macchia di vino porto [7].
Neurologico e oftalmologico I segni della sindrome di Sturge-Weber sono progressivo e di solito sviluppare nei primi due anni di vita. Questi possono includere:
- Convulsioni e l'epilessia
- Emiparesi ed eventi simili all'ictus
- Problemi di comportamento
- Difetti del campo visivo e glaucoma
- Carenza di ormone della crescita.
Quali sono le complicazioni della sindrome di Sturge-Weber?
Le complicanze della sindrome di Sturge-Weber dipendono dall'entità delle malformazioni vascolari e da altre caratteristiche cliniche. Sono estremamente variabili.
Come viene diagnosticata la sindrome di Sturge-Weber?
La diagnosi della sindrome di Sturge-Weber si basa sul riscontro delle caratteristiche macchie di vino porto trigeminale e delle malformazioni leptomeningee capillare-venose. Una diagnosi basata sulla leptomeningea lesioni dipende solo da sviluppo sintomi.
Se ci sono sintomi o risultati neurologici, risonanza magnetica (Risonanza magnetica) del cervello viene eseguita con contrasto gadolinio per rilevare malformazioni leptomeningee capillare-venose.
Qual è diagnosi differenziale per la sindrome di Sturge-Weber?
Le caratteristiche di altre sindromi da malformazione vascolare sono descritte di seguito.
- La sindrome di Klippel-Trénaunay presenta malformazioni capillari più estese, colpisce le estremità e il tronco ed è associata a ipertrofia dell'arto colpito.
- La sindrome di Parkes-Weber si presenta con un'ampia malformazione capillare in un arto e ipertrofia dell'arto colpito. Esistono più shunt artero-venosi a flusso rapido.
- La sindrome di Servelle-Martorell, una rara malattia angiodisplastica congenita, è associata a ipotrofia progressiva delle estremità.
- La sindrome di Proteus si presenta con asimmetrico e una crescita eccessiva sproporzionata di parti del corpo. La sindrome è il risultato di una mutazione attivante somatica nel AKT1 oncogene.
- La sindrome di CLOVES si presenta con crescita eccessiva lipomatosa congenita, malformazione vascolare, epidermico nevo, anomalie spinali / scheletriche /scoliosi. La sindrome deriva dall'attivazione del mosaico somatico mutazioni a PIK3CA gene.
Qual è il trattamento per la sindrome di Sturge-Weber?
Non esiste un trattamento specifico per la sindrome di Sturge-Weber. Il trattamento consiste nella gestione del cutaneo, neurologici e oculare sintomi, con scarso successo.
Il trattamento delle crisi epilettiche in pazienti con sindrome di Sturge-Weber con farmaci antiepilettici non è sempre efficace [8]. Nessun trattamento singolo sembra esserlo più alto per gli altri.
Le macchie di vino porto possono essere trattate con tintura pulsata Essere con risultati variabili [9]. A causa dei risultati generalmente scarsi della tintura pulsata laser solo, attuale gli agenti antiangiogenici sono in fase di sperimentazione come allegato terapie.
Poiché il glaucoma è una complicanza comune della sindrome di Sturge-Weber, controlli regolari della vista con a oculista sono consigliati. Il glaucoma viene trattato chirurgicamente e clinicamente [10].
I bambini con diagnosi di sindrome di Sturge-Weber dovrebbero essere trattati con aspirina a basso dosaggio [11,12]. La terapia antitrombotica può prevenire la progressione della malattia, che può influenzare il flusso sanguigno al cervello e portare a danni neuronali.
Qual è il risultato della sindrome di Sturge-Weber?
il previsione La sindrome di Sturge-Weber dipende dal grado di coinvolgimento del cervello e della pelle. Le macchie estese di vino porto sono associate ad un aumentato rischio di epilessia e glaucoma, mentre bilaterale Le malformazioni vascolari leptomeningee sono associate a disabilità intellettive e di apprendimento. [13]. L'insorgenza di crisi prima di un anno ha un effetto significativo sulla funzione cognitiva e motoria nei bambini con sindrome di Sturge-Weber [14].
Si stima che circa un adulto su due con sindrome di Sturge-Weber abbia difetti neurologici, anche in quelli che inizialmente erano asintomatico.