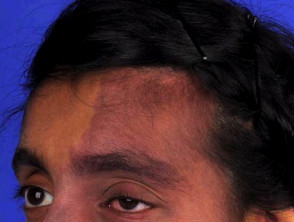
Entendiendo el Síndrome de Sturge-Weber: Causas, Síntomas y Características
El síndrome de Sturge-Weber es un trastorno neurocutáneo poco frecuente, que es congénito pero no hereditario. Se distingue por la presencia de una malformación capilar en la piel del rostro, comúnmente conocida como mancha de vino de Oporto, junto con malformaciones venosas capilares en el cerebro y los ojos [1].
Síndrome de Sturge-Weber Definición
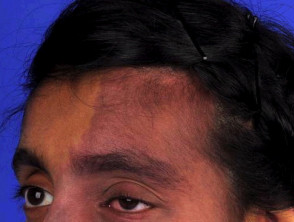
Síndrome de Sturge-Weber
Prevalencia y Naturaleza del Síndrome de Sturge-Weber
Se estima que la incidencia del síndrome de Sturge-Weber afecta a aproximadamente uno de cada 20,000 a 50,000 recién nacidos [2]. Debido a que es provocado por una somático mosaico mutación, esta condición no se transmite genéticamente de padres a hijos.
Causas Genéticas del Síndrome de Sturge-Weber
La etiología del síndrome de Sturge-Weber reside en una mutación somática en mosaico del gene GNAQ, ubicado en el cromosoma 9 [3]. Esta alteración genética ocurre en las células del cuerpo después de la fertilización y la formación del cigoto. El gen GNAQ es fundamental en la regulación de las vías de señalización intracelular, y su mutación provoca una proliferación o maduración inadecuada de los capilares en las células afectadas.
Manifestaciones Clínicas del Síndrome de Sturge-Weber
El síndrome de Sturge-Weber se caracteriza por malformaciones vasculares evidentes en la cara, los ojos y el cerebro. Estos signos son observables desde el nacimiento.
- Las manchas de vino de Oporto constituyen la malformación vascular más frecuente. Afectan alrededor de tres de cada 1000 bebés, aunque la mayoría de ellas no están relacionadas con el síndrome de Sturge-Weber [4].
- Cuando están asociadas al síndrome, estas manchas se localizan típicamente en la distribución del nervio trigémino, específicamente en la primera y segunda división, afectando la frente y el párpado superior [5]. Es importante notar que también pueden presentarse en ambos lados del rostro [6].
- Surgen malformaciones vasculares leptomeníngeas en el cerebro, ubicadas en el mismo lado que la mancha facial.
- En algunos casos, estas malformaciones vasculares leptomeníngeas pueden manifestarse sin la presencia de una mancha de vino de Oporto concomitante [7].
Neurológico y oftalmológico Los signos asociados al síndrome de Sturge-Weber tienden a ser progresivos y suelen desarrollar durante los primeros dos años de vida. Dichas manifestaciones pueden incluir:
- Convulsiones
- Epilepsia
- Hemiparesia y episodios similares a un accidente cerebrovascular
- Alteraciones del comportamiento
- Defectos del campo visual y glaucoma
- Déficit en la hormona del crecimiento.
Complicaciones Asociadas al Síndrome de Sturge-Weber
Las complicaciones que surgen del síndrome de Sturge-Weber están directamente ligadas a la extensión de las distintas malformaciones vasculares y a otros signos clínicos. Es importante destacar que su manifestación es notablemente variable entre los afectados.
Métodos de Diagnóstico para el Síndrome de Sturge-Weber
El diagnóstico del síndrome de Sturge-Weber se establece principalmente al identificar la característica mancha en vino de Oporto del trigémino junto con la presencia de malformaciones capilares-venosas leptomeningeas. Si las lesiones leptomeningeas son el único indicador, el diagnóstico dependerá de la aparición de síntomas clínicos asociados a su desarrollo.
En presencia de signos neurológicos o síntomas evidentes, se procede a solicitar una imagen de resonancia magnética (Resonancia magnética) cerebral, utilizando contraste de gadolinio para confirmar y localizar las malformaciones capilares-venosas leptomeningeas.
Diagnóstico Diferencial en el Síndrome de Sturge-Weber
Es fundamental distinguir el síndrome de Sturge-Weber de otras condiciones que presentan malformaciones vasculares. A continuación, se detallan las características distintivas de síndromes relacionados:
- El síndrome de Klippel-Trénaunay
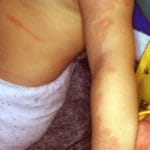 Entendiendo el Síndrome de Klippel-Trénaunay: Definición y Características El síndrome de Klippel-Trénaunay (KTS) es un congénito vascular óseo poco frecuente, caracterizado por una tríada distintiva de manifestaciones clínicas que incluyen: Capilar malformación (también conocida como mancha de vino de Oporto) Venoso malformación (por ejemplo, presencia de venas varicosas extensas) Miembro hipertrofia (aumento de tamaño, afectando generalmente una sola extremidad) [1,2]. Este trastorno también es reconocido bajo los nombres de síndrome más involucra malformaciones capilares más extensas, afectando predominantemente extremidades y tronco, y se asocia frecuentemente con hipertrofia de la extremidad afectada.
Entendiendo el Síndrome de Klippel-Trénaunay: Definición y Características El síndrome de Klippel-Trénaunay (KTS) es un congénito vascular óseo poco frecuente, caracterizado por una tríada distintiva de manifestaciones clínicas que incluyen: Capilar malformación (también conocida como mancha de vino de Oporto) Venoso malformación (por ejemplo, presencia de venas varicosas extensas) Miembro hipertrofia (aumento de tamaño, afectando generalmente una sola extremidad) [1,2]. Este trastorno también es reconocido bajo los nombres de síndrome más involucra malformaciones capilares más extensas, afectando predominantemente extremidades y tronco, y se asocia frecuentemente con hipertrofia de la extremidad afectada. - El síndrome de Parkes-Weber se caracteriza por una gran mancha capilar en una extremidad junto con hipertrofia de dicha extremidad, presentando además múltiples derivaciones arteriovenosas de alto flujo.
- El síndrome de Servelle-Martorell, una rara enfermedad angiodisplásica congénita, se asocia con una hipotrofia progresiva de las extremidades.
- El síndrome de ProteusComprensión del Síndrome de Proteus: Síntomas, Causas y Características El síndrome de Proteus, también conocido como síndrome de Elattoproteus o enfermedad del hombre elefante, es una condición medicalmente fascinante. Esta enfermedad ganó notoriedad a través de la película "El hombre elefante", que retrataba la vida de Joseph Merrick. Si bien en el pasado se creyó que Merrick padecía neurofibromatosis, hoy se considera que probablemente sufrió una manifestación severa del síndrome más se manifiesta por un crecimiento corporal marcadamente asimétrico y desproporcionado, resultado directo de una mutación somática activadora en el oncogén AKT1.
- El síndrome de CLOVES
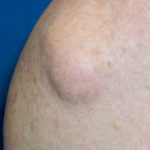 Entendiendo el Síndrome de CLOVES: Causas, Características y Criterios Diagnósticos El síndrome de CLOVES es un trastorno metabólico congénito y progresivo poco frecuente que impacta crucialmente múltiples sistemas orgánicos. Esta condición afecta significativamente la piel, el sistema vascular y la estructura musculoesquelética del individuo [1]. La nomenclatura "CLOVES" funciona como un mnemónico útil, ya que describe los principales componentes clínicos del síndrome: • Crecimiento lipomatoso Congenital • Malformaciones Vasculares (Variaciones) más se identifica por sobrecrecimiento lipomatoso congénito, malformaciones vasculares, presencia de nevo epidérmico, y anomalías espinales/esqueléticas, incluyendo escoliosis. Este síndrome es causado por mutaciones somáticas de activación en el gen PIK3CA.
Entendiendo el Síndrome de CLOVES: Causas, Características y Criterios Diagnósticos El síndrome de CLOVES es un trastorno metabólico congénito y progresivo poco frecuente que impacta crucialmente múltiples sistemas orgánicos. Esta condición afecta significativamente la piel, el sistema vascular y la estructura musculoesquelética del individuo [1]. La nomenclatura "CLOVES" funciona como un mnemónico útil, ya que describe los principales componentes clínicos del síndrome: • Crecimiento lipomatoso Congenital • Malformaciones Vasculares (Variaciones) más se identifica por sobrecrecimiento lipomatoso congénito, malformaciones vasculares, presencia de nevo epidérmico, y anomalías espinales/esqueléticas, incluyendo escoliosis. Este síndrome es causado por mutaciones somáticas de activación en el gen PIK3CA.
Opciones de Tratamiento para el Síndrome de Sturge-Weber
Actualmente, no existe un tratamiento curativo específico para el síndrome de Sturge-Weber. El enfoque terapéutico se centra en el manejo sintomático de las manifestaciones cutáneas, neurológicas y oculares, aunque el éxito suele ser limitado.
El control de las convulsiones mediante fármacos antiepilépticos en pacientes con Sturge-Weber no siempre resulta efectivo [8]. De hecho, ningún abordaje terapéutico único ha demostrado ser notablemente superior a los demás.
Las manchas en vino de Oporto pueden ser tratadas con láser de colorante pulsado, reportándose resultados variables [9]. Dado que los resultados del tratamiento con solo láseres de colorante pulsado suelen ser modestos, se están evaluando agentes antiangiogénicos como adyuvantes o terapias actuales.
Puesto que el glaucoma es una complicación frecuente, se aconsejan revisiones oftalmológicas periódicas con un oftalmólogo. El glaucoma, en caso de desarrollarse, requiere manejo tanto médico como quirúrgico [10].
Además, se recomienda administrar aspirina en dosis bajas a los lactantes que son diagnosticados con el síndrome de Sturge-Weber [11,12]. Esta terapia antitrombótica tiene el potencial de prevenir la progresión de la enfermedad, la cual podría comprometer el flujo sanguíneo cerebral y derivar en daño neuronal.
Pronóstico en el Síndrome de Sturge-Weber
El pronóstico del síndrome de Sturge-Weber está intrínsecamente definido por el grado de afectación tanto cerebral como cutánea. Las manchas extensas en vino de Oporto se correlacionan con un riesgo incrementado de desarrollar epilepsia y glaucoma. Por otro lado, las malformaciones vasculares leptomeningeas bilaterales se asocian con secuelas de discapacidad intelectual y dificultades de aprendizaje [13]. La aparición temprana de convulsiones (antes del primer año de vida) impacta significativamente la función cognitiva y motora en los niños afectados por Sturge-Weber [14].
Se estima que aproximadamente la mitad de los adultos que padecen el síndrome de Sturge-Weber presentarán secuelas neurológicas, incluso aquellos que inicialmente se mantuvieron asintomáticos durante las primeras etapas.


