La classificazione internazionale 2017 delle sindromi di Ehlers-Danlos
Vedi Ehlers - Danlos sindrome.
Di seguito sono descritti i sottotipi specifici della sindrome di Ehlers-Danlos (EDS) o delle sindromi di Ehlers-Danlos. [1,2]. Loro includono:
- Sindrome di Ehlers-Danlos classica (cEDS)
- Sindrome di Ehlers-Danlos classica (clEDS)
- Sindrome cardiaco-valvolare di Ehlers-Danlos (cvEDS)
- Vascolare Sindrome di Ehlers-Danlos (vEDS)
- Sindrome di Ehlers-Danlos ipermobile (hEDS)
- Arthrochalasia sindrome di Ehlers-Danlos (aEDS)
- Dermatosparaxis sindrome di Ehlers-Danlos (dEDS)
- Sindrome cifoscoliotica di Ehlers-Danlos (kEDS)
- Fragile cornea sindrome (BCS)
- Sindrome spondilodisplastica di Ehlers-Danlos (spEDS)
- Sindrome muscoloscheletrica di Ehlers-Danlos (mcEDS)
- Sindrome miopatica di Ehlers-Danlos (mEDS)
- Sindrome parodontale di Ehlers-Danlos (pEDS)
Sindrome di Ehlers-Danlos
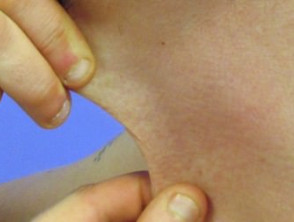
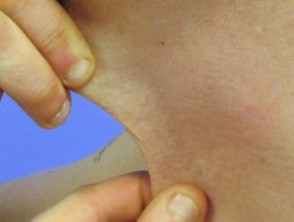
Iperelasticità della pelle del collo
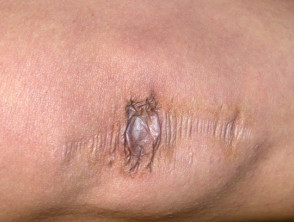
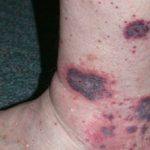
Cicatrici estese in un paziente con sindrome di Ehlers-Danlos.
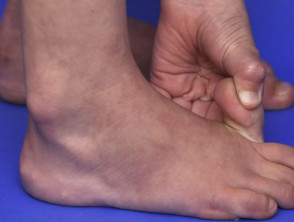
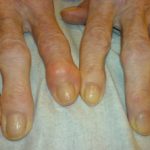
Articolazioni ipermobile
Guarda altre immagini della sindrome di Ehlers-Danlos.
Sindrome di Ehlers-Danlos classica (cEDS)
Il modello di ereditarietà cEDS è autosomica dominante.
- Gli affetti geni in cEDS lo sono COL5A1 (MIM 130000), COL5A2, (MIM 130010) e COL1A1 che interessano il tipo V e il tipo I. collagene.
- Il 50% dei pazienti con diagnosi di cEDS ha a de-novo mutazione (cioè, nessuno dei genitori è interessato).
- La gravità varia in modo significativo, anche all'interno della stessa famiglia.
- Vascolare fragilità può includere dissezione spontanea o rottura delle mediane arterie.
- Di solito i pazienti con cEDS hanno un'iperestensibilità cutanea marcata e allargata atrofica cutaneo cicatrici e molto diffuso ipermobilità articolare.
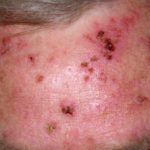
- Altre caratteristiche di cEDS sono emosiderina dichiarazione (particolarmente evidente sugli stinchi e sul dorso delle mani), sottocutaneo sferoidi (lobi di grasso calcificati dopo la perdita di afflusso di sangue) e pseudotumori dei molluschi (pelle carnosa ispessita lesioni/ cicatrici su ginocchia e gomiti).
- Non ci sono smagliature (smagliature) nei cEDS.
- Elettrone tipico microscopia i risultati sono "fiori di collagene" o "cavolfiori" a causa di fibrillogenesi anormale.
- Le complicanze della cEDS sono le valvole cardiache deformità, dilatazione dell'aorta e aortico dissezione
Sindrome di Ehlers-Danlos classica (clEDS)
Il modello di ereditarietà di clEDS è autosomica recessiva.
- Gli affetti gene in clEDS lo è TNXB che interessano il proteina Tenascin XB (MIM 606408)
- I pazienti con ClEDS hanno ipermobilità articolare generalizzata, pelle morbida e vellutata, iperestensibile e gravi lividi.
- Non ci sono cicatrici atrofiche su CLEDS.
Sindrome cardiaco-valvolare di Ehlers-Danlos (cvEDS)
Il pattern di ereditarietà cvEDS è autosomica recessiva.
- Il gene interessato in cvEDS è COL1A2 che influenzano la proteina collagene di tipo I (MIM 225320)
- I pazienti con cvEDS hanno lievi segni di EDS ma hanno un grave difetto aortico / coinvolgimento della valvola cardiaca che richiede un intervento chirurgico.
Sindrome vascolare di Ehlers-Danlos (vEDS)
Il modello di ereditarietà vEDS è autosomica dominante.
- I geni colpiti in vEDS sono COL3A1 che interessano il collagene di tipo III e COL1A1 che colpisce il collagene di tipo 1 (MIM 130050)
- Il 50% dei pazienti con diagnosi di vEDS ha a de novo mutazione (cioè, nessuno dei genitori è interessato).
- il mediano l'aspettativa di vita è di 50 anni.
- Le caratteristiche cliniche sottili o assenti della vEDS in alcuni pazienti rendono questo sottotipo difficile da diagnosticare, con la morte improvvisa tra i 30 ei 40 anni che è l'unica caratteristica presente.
- I VEDS possono essere identificati alla nascita se è presente un evidente piede torto deformità o lussazione dell'anca.
- Le caratteristiche dei vEDS sono lividi gravi e sottili traslucido aspetto caratteristico della pelle e del viso (guance tese e infossate, labbra e naso sottili e occhi prominenti).
- Le complicazioni del vEDS sono arterioso dissezione (inclusa l'aorta), rottura (inclusa la rottura dell'intestino, con il colon sigmoideo che è il sito più comune) e complicanze ostetriche (rottura uterina, massiccia postpartum emorragia).
Sindrome di Ehlers-Danlos ipermobile (hEDS)
Il modello di ereditarietà della hEDS è autosomica dominante.
- Il gene interessato in hEDS (MIM 130020) è sconosciuto.
- I pazienti con hEDS non sono soggetti a complicazioni potenzialmente letali, ma i loro effetti possono essere debilitanti.
- Le caratteristiche dell'hEDS sono ipermobilità articolare generalizzata, ipermobilità spinale e frequenti lussazioni o sublussazioni articolari (spalla, rotula o articolazione temporo-mandibolare).
- Non ci sono pseudotumori di molluschi.
- Le complicanze dell'hEDS sono la dilatazione della radice aortica, autonomo disfunzione (ortostatica posturale tachicardia sindrome), grave cronica dolori articolari scoliosie prematuro artrosi.
Arthrochalasia sindrome di Ehlers-Danlos (aEDS)
Il modello di ereditarietà degli aEDS è autosomica dominante.
- I geni interessati negli aEDS sono COL1A1 e COL1A2 che colpisce il collagene di tipo I (MIM 130060).
- Come la parola descrive, artro- significa articolazione e -calasia significa rilassamento; la mutazione si traduce in un collagene debole anormale.
- I pazienti con DAE corrono un rischio per tutta la vita simultaneo lussazione di più articolazioni principali.
- Le caratteristiche degli aEDS sono ipotonia, grave ipermobilità articolare generalizzata, congenita bilaterale lussazione dell'anca e osteopenia.
Dermatosparaxis sindrome di Ehlers-Danlos (dEDS)
Il pattern di ereditarietà della dEDS è autosomica recessiva.
- Il gene interessato in dEDS è ADAMTS2 che influenzano la proteina ADAMTS-2 (MIM 225410).
- Sparaxis deriva da una parola greca che significa "strappare".
- Le caratteristiche dei dEDS sono grave fragilità cutanea, cute ridondante lassista, chiusura ritardata delle fontanelle, colore blu sclera, viscerale fragilità e ritardo della crescita postnatale con conseguente bassa statura.
- Ci sono caratteristiche geroglifiche fibrille' in microscopio elettronico della pelle.
Sindrome cifoscoliotica di Ehlers-Danlos (kEDS)
Il modello di ereditarietà di kEDS è autosomica recessiva.
- I geni colpiti da dEDS lo sono PLOD1 che influenzano la proteina LH1 (MIM 225400) e FKBP14 che influenzano la proteina FKBP22 (MIM 614557).
- Gene mutazioni provocare una carenza di lisilidrossilasi, che è un modificatore del collagene enzima importante formare legami incrociati tra trimeri di collagene per aumentare la forza del collagene.
- kEDS è caratterizzato da un inizio precoce progressivo Cifoscoliosi, ipotonia, ritardo motorio grossolano, sclera blu con pressione intraoculare elevata, ipermobilità articolare generalizzata e facile formazione di lividi.
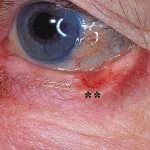
Sindrome corneale fragile (BCS)
Il modello di ereditarietà di BCS è autosomica recessiva.
- I geni colpiti da dEDS lo sono PRDM5 che influenzano la proteina PRDM5 (MIM 614170) e ZNF469 che interessano ZNF469 (MIM 229200).
- I pazienti con BCS hanno una cornea sottile e fragile che può pugno fuori spontaneamente o dopo minore trauma.
- Le caratteristiche della BCS sono cornea sottile, cheratocono, sclera blu, rischio di rottura del globo, distacco della retina e miopia elevata (miopia).
Sindrome spondilodisplastica di Ehlers-Danlos (spEDS)
Il modello di ereditarietà di spEDS è autosomica recessiva.
- I geni colpiti da spEDS sono B4GALT7 che influenzano la proteina beta4GalT7 (MIM 130070), B3GALT6 che interessano beta3GalT6 (MIM 615349) e SLC39A13 che interessa ZIP13 (MIM 612350).
- Le caratteristiche di spEDS sono bassa statura, ipotonia, piegamento degli arti, osteopenia e pelle iperestensibile, sottile e pastosa.
Sindrome muscoloscheletrica di Ehlers-Danlos (mcEDS)
Il modello di ereditarietà di mcEDS è autosomica recessiva.
- I geni colpiti da mcEDS lo sono CHST14 che influenzano la proteina D4ST1 (MIM 601776) e DSE che interessano DSE (MIM 615539).
- Le caratteristiche di mcEDS sono congenite contratture con conseguente piede torto, ricorrente lussazioni, pollici addotti (stretti) durante l'infanzia, pelle iperestensibile, lividi facili, pelle fragile e cicatrici atrofiche.
Sindrome miopatica di Ehlers-Danlos (mEDS)
I modelli di ereditarietà dei mEDS possono essere autosomici dominanti o autosomici recessivi.
- Il gene interessato in mEDS è COL12A1 che colpisce il collagene di tipo XII (MIM 616471).
- La debolezza muscolare presente nell'infanzia e nell'infanzia tende a migliorare nell'età adulta, con un certo deterioramento dopo i 40 anni.
Sindrome parodontale di Ehlers-Danlos (pEDS)
Il modello di ereditarietà della pEDS è autosomica dominante.
- I geni colpiti da pEDS sono C1R che influenzano la proteina C1r e C1S che interessano C1 (MIM 130080).
- Le caratteristiche della pEDS sono gravi con esordio precoce parodontite (gonfiore delle gengive rosse, recessione gengivale, perdita precoce dei denti), livido giallastro pretibiale piatti, ipermobilità articolare e pelle iperestensibile.