Che cos'è gioventù sistemico granulomatosi?
La granulomatosi sistemica giovanile è un nuovo termine proposto per includere Blau sindrome (MIM186580, chiamata anche sindrome di Jabs) e sarcoidosi ad esordio precoce (MIM609464), poiché queste due condizioni sono state ora collegate dopo l'identificazione di mutazioni influenzando lo stesso gene e molecolare base. Pediatrico granulomatoso artrite È stato anche proposto come nome unificante, ma questo si concentra solo sulla caratteristica clinica più comune, che non si verifica in tutti i pazienti.
La granulomatosi sistemica giovanile è una delle monogenico autoinfiammatorio sindromi.
Che cosa causa la granulomatosi sistemica giovanile e chi la prende?
La granulomatosi sistemica giovanile è il risultato di mutazioni nel dominio NACHT del gene CARD15 (dominio di reclutamento delle caspasi 15), noto anche come gene NOD2 situato in cromosoma 16q12.1-13. Fino ad ora questo è sempre stato uno eterozigote singolonucleotide sostituzione. R224W e R334Q sono le mutazioni più comuni riportate. Le stesse mutazioni CARD15 sono state identificate sia nella sindrome di Blau che nella sarcoidosi ad esordio precoce, ma non nella sarcoidosi adulta. Ad oggi, tutti i pazienti che ne hanno mutazione hanno mostrato segni clinici di questa sindrome (100% penetranza).
CARD15 codifica il NOD2 (Nod2) proteina ed è espresso principalmente in citoplasma di monociti. NOD2 riconosce il muramil dippepetide nella parete cellulare gram-negativa e gram-positiva. batteri. Il legame risulta nell'attivazione di NK-KB e nel meccanismo di infiammazione non implica eccesso interleuchina(IL) -1 attività. Le mutazioni del gene CARD15 danno luogo a un singolo aminoacidi cambiamento nella proteina NOD2.
La sindrome di Blau è stata definita come a autosomica condizione dominante, mentre la sarcoidosi ad esordio precoce non lo era famiglia. Tuttavia, ora sembra che la sarcoidosi ad esordio precoce sia dovuta a una mutazione sporadica, rispetto alla mutazione ereditaria nella sindrome di Blau. Una donna con diagnosi di sarcoidosi a esordio precoce a causa di una mancanza di storia familiare ha successivamente dato alla luce un bambino affetto, per il quale la diagnosi era la sindrome di Blau.
La granulomatosi sistemica giovanile si manifesta nella prima infanzia, prima dei 4 anni. È strano, con a incidenza tasso inferiore a 1 per 1,5 milioni di persone-anno. La sarcoidosi ad esordio precoce colpisce i bianchi più spesso dei neri, in contrasto con la sarcoidosi ad esordio nell'adulto.
Caratteristiche cliniche della granulomatosi sistemica giovanile
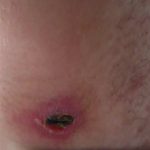
Esiste un coinvolgimento sequenziale della pelle, delle articolazioni e degli occhi nella granulomatosi sistemica giovanile, che di solito inizia prima dei 3 anni di età. Non tutti i pazienti sviluppare l'intera triade classica nel tempo. Approssimativamente il 40% svolge tutte e tre le funzioni. La variabilità nella presentazione clinica si verifica anche all'interno di una famiglia.
| Pelle lesioni |
|---|
|
| Articolazioni |
|
| Occhi |
|
| Altro dimostrazioni |
|
- Il coinvolgimento degli organi interni è raro e, in particolare, polmonare o ilare. linfonodo non si verifica ingrossamento, a differenza della sarcoidosi ad esordio nell'adulto.
- Se non trattato, l'80% sviluppa gravi disturbi visivi e articolari. deformità.
- Una maggiore gravità è stata segnalata nelle generazioni successive ("anticipazione").
Come viene diagnosticata la granulomatosi sistemica giovanile?
Pelle biopsia campione granulomatoso non caseoso dermatite. Questo dermatopatologia termine descrive il particolare schema di miscelazione infiammatorio infiltrarsi caratteristica della sarcoidosi. il granulomi sono in cima derma e si trovano spesso intorno ai follicoli piliferi. No-involucro Si vedono anche granulomi biopsie dall'occhio, Sì fidanzato delle articolazioni colpite e altri siti colpiti.
I raggi X delle articolazioni colpite sono quasi normali e non mostrano il erosivo cambiamenti nell'artrite reumatoide giovanile.
L'esame della vista con lampada a fessura deve essere eseguito regolarmente poiché la malattia dell'occhio precoce può essere asintomatica.
Gli studi di mutazione del gene CARD15 (NOD2) possono essere eseguiti in a membrana mucosa raschiato via.
Qual è il trattamento per la granulomatosi sistemica giovanile?
Genetico È necessaria una consulenza e prenatale La diagnosi può essere presa in considerazione se la mutazione è stata identificata nel probando.
Per prevenire danni agli occhi, è necessaria l'immunosoppressione sistemica a lungo termine. Le opzioni includono:
- sistemico corticosteroidi
- metotrexato
- ciclosporina
- micofenolato mofetile
- talidomide
- tumore necrosi fattore inibitori (infliximab, etanercept, adalimumab)
- antagonisti dell'interleuchina-1 come anakinra
L'infiammazione degli occhi può essere la più difficile manifestazione trattare e la principale causa di complicanze a lungo termine.