Comprensión de la Granulomatosis Sistémica Juvenil: Causas y Características
La **Granulomatosis Sistémica Juvenil** es una denominación recientemente propuesta que engloba el Síndrome de Blau (MIM186580, también conocido como Síndrome de Jabs) y la sarcoidosis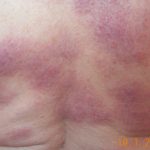 Sarcoidosis: Definición, Riesgos y Enfermedades Desencadenantes La sarcoidosis se define como un trastorno inflamatorio multisistémico, caracterizado por la formación de granulomas. Estos son cúmulos localizados de células inflamatorias que pueden afectar diversas partes del cuerpo. Desde una perspectiva patológica, estos granulomas se describen típicamente como epitelioides no caseificantes, lo que los diferencia claramente de los granulomas caseificantes asociados a dolencias como la tuberculosis. Usualmente, esta condición se origina en los más de inicio temprano (MIM609464). Esta unificación se debe al descubrimiento de mutaciones idénticas que afectan el mismo gen y comparten una base molecular común. Aunque también se ha sugerido el término Artritis granulomatoso Pediátrico, esta opción prioriza solo la manifestación clínica más frecuente, la cual no está presente en todos los casos.
Sarcoidosis: Definición, Riesgos y Enfermedades Desencadenantes La sarcoidosis se define como un trastorno inflamatorio multisistémico, caracterizado por la formación de granulomas. Estos son cúmulos localizados de células inflamatorias que pueden afectar diversas partes del cuerpo. Desde una perspectiva patológica, estos granulomas se describen típicamente como epitelioides no caseificantes, lo que los diferencia claramente de los granulomas caseificantes asociados a dolencias como la tuberculosis. Usualmente, esta condición se origina en los más de inicio temprano (MIM609464). Esta unificación se debe al descubrimiento de mutaciones idénticas que afectan el mismo gen y comparten una base molecular común. Aunque también se ha sugerido el término Artritis granulomatoso Pediátrico, esta opción prioriza solo la manifestación clínica más frecuente, la cual no está presente en todos los casos.
En esencia, la granulomatosis sistémica juvenil se clasifica como uno de los síndromes autoinflamatorio monogénico que afectan al organismo.
Causas Genéticas y Epidemiología de la Granulomatosis Sistémica Juvenil
El origen de la granulomatosis sistémica juvenil reside en las mutaciones localizadas en el dominio NACHT del gen CARD15 (dominio 15 de reclutamiento de caspasas), también conocido como gen NOD2. Este gen se localiza en el cromosoma 16q12.1-13. Hasta la fecha, las alteraciones detectadas han sido siempre sustituciones de un solo nucleótido heterocigoto en el gen. Las variantes R224W y R334Q son consistentemente las mutaciones más frecuentes reportadas en la literatura médica.
Es crucial destacar que estas mismas mutaciones en CARD15 se han identificado tanto en el síndrome de Blau como en la sarcoidosis de inicio temprano, pero no se encuentran en la sarcoidosis que se desarrolla en la edad adulta. Con una penetrancia del 100%, todos los pacientes que portan dicha mutación han manifestado signos clínicos de este síndrome.
El gen CARD15 es el responsable de codificar la proteína NOD2, la cual se expresa predominantemente en el citoplasma de los monocitos. La proteína NOD2 tiene la función de reconocer el muramil dipéptido presente en la pared celular de bacterias tanto gramnegativas como grampositivas. Tras esta unión, se activa la vía NK-κB, y el mecanismo de inflamación subsiguiente no involucra un exceso de actividad de la interleucina (IL)-1. Las mutaciones en el gen CARD15 resultan en un cambio de un solo aminoácidos en la estructura de la proteína NOD2.
Tradicionalmente, el síndrome de Blau se clasificaba como una condición dominante autosómica, mientras que la sarcoidosis de inicio temprano no se consideraba familiar. No obstante, la evidencia actual sugiere que la sarcoidosis de inicio temprano se debe a una mutación esporádica, a diferencia de la mutación hereditaria observada en el síndrome de Blau. Un ejemplo ilustrativo fue el de una paciente diagnosticada con sarcoidosis de inicio temprano por ausencia de antecedentes familiares, quien posteriormente dio a luz a un bebé afectado, reclasificando su diagnóstico original a síndrome de Blau.
La granulomatosis sistémica juvenil se manifiesta típicamente durante la primera infancia, antes de los 4 años de edad. Es una condición rara, con una tasa de incidencia inferior a 1 caso por 1.5 millones de personas-año. Además, la sarcoidosis de inicio temprano afecta con mayor prevalencia a individuos de raza blanca en comparación con la población negra, lo que difiere del patrón observado en la sarcoidosis que aparece en la edad adulta.
Características Clínicas Clave de la Granulomatosis Sistémica Juvenil
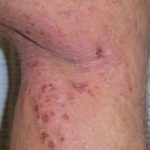
La granulomatosis sistémica juvenil (GSJ) exhibe una afectación secuencial que típicamente involucra la piel, las articulaciones y los ojos, con una edad de inicio que generalmente precede a los 3 años. Es importante notar que no todos los pacientes **desarrollar**án la tríada sintomática clásica a lo largo del tiempo; solo aproximadamente el 40% la manifiesta completamente. La presentación clínica es notablemente variable, incluso entre miembros de la misma familia.
| Lesiones Cutáneas |
|---|
|
| Afectación Articular |
|
| Compromiso Ocular |
|
| Otras Manifestaciones |
|
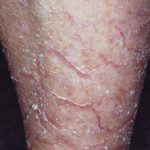

La presentación clínica de la granulomatosis sistémica juvenil es compleja y heterogénea, requiriendo observación detallada para identificar la tríada clásica de afectación cutánea, articular y ocular. Dada la cronicidad de muchas de estas **manifestaciones**, el seguimiento continuo es esencial para manejar complicaciones como el glaucoma o las secuelas cutáneas.
- Es poco frecuente la afectación de órganos internos, y el agrandamiento del ganglio linfático hiliar o pulmonar no suele ocurrir, a diferencia de la sarcoidosis que comienza en la edad adulta.
- Si no se administra tratamiento, el 80% de los pacientes desarrollan graves alteraciones visuales y deformidades articulares.
- Se ha documentado una mayor severidad en las generaciones posteriores del trastorno, un fenómeno conocido como "anticipación".
Diagnóstico de la Granulomatosis Sistémica Juvenil
La biopsia de piel revela una dermatitis Comprendiendo la Dermatitis: Definición, Causas y Tipos Comunes La dermatitis abarca un conjunto de afecciones inflamatorias que se manifiestan a través de cambios específicos en la epidermis, manifestándose frecuentemente como picazón intensa. Esta condición es notablemente común, afectando a cerca de una quinta parte de la población en algún momento de sus vidas. Debido a su etiología diversa, la dermatitis presenta múltiples patrones de manifestación clínica. Los términos "dermatitis" y más granulomatosa no caseificante. Este término de dermatopatología describe el patrón específico de infiltrado inflamatorio característico de la sarcoidosis. Los granulomas se sitúan en la parte superior de la dermis y frecuentemente rodean los folículos pilosos. Los granulomas no-caseating también se observan en biopsias tomadas del ojo, del sinovio de las articulaciones afectadas y de otros sitios comprometidos.
Comprendiendo la Dermatitis: Definición, Causas y Tipos Comunes La dermatitis abarca un conjunto de afecciones inflamatorias que se manifiestan a través de cambios específicos en la epidermis, manifestándose frecuentemente como picazón intensa. Esta condición es notablemente común, afectando a cerca de una quinta parte de la población en algún momento de sus vidas. Debido a su etiología diversa, la dermatitis presenta múltiples patrones de manifestación clínica. Los términos "dermatitis" y más granulomatosa no caseificante. Este término de dermatopatología describe el patrón específico de infiltrado inflamatorio característico de la sarcoidosis. Los granulomas se sitúan en la parte superior de la dermis y frecuentemente rodean los folículos pilosos. Los granulomas no-caseating también se observan en biopsias tomadas del ojo, del sinovio de las articulaciones afectadas y de otros sitios comprometidos.
Las radiografías de las articulaciones afectadas suelen ser normales y no muestran los cambios erosivos típicos de la artritis reumatoide Artritis Reumatoide: Definición y Manifestaciones Cutáneas La artritis reumatoide (AR) es una compleja enfermedad clasificada dentro de los trastornos del tejido conectivo. Se define como un crónico trastorno sistémico de naturaleza inflamatoria que progresa dañando progresivamente las articulaciones del cuerpo humano. Los síntomas cardinales de la AR incluyen dolor articular, hinchazón, deformidad, debilidad y una marcada rigidez. Estos efectos suelen concentrarse en las articulaciones más pequeñas, como las de manos, más juvenil.
Artritis Reumatoide: Definición y Manifestaciones Cutáneas La artritis reumatoide (AR) es una compleja enfermedad clasificada dentro de los trastornos del tejido conectivo. Se define como un crónico trastorno sistémico de naturaleza inflamatoria que progresa dañando progresivamente las articulaciones del cuerpo humano. Los síntomas cardinales de la AR incluyen dolor articular, hinchazón, deformidad, debilidad y una marcada rigidez. Estos efectos suelen concentrarse en las articulaciones más pequeñas, como las de manos, más juvenil.
Es crucial realizar un examen ocular regular con lámpara de hendidura, ya que la enfermedad ocular en sus etapas iniciales puede ser completamente asintomática.
Los estudios de mutación del gen CARD15 (NOD2) pueden realizarse a partir de un raspado de mucosa.
Tratamiento para la Granulomatosis Sistémica Juvenil
Genético Se recomienda el asesoramiento y, si la mutación ha sido confirmada en el paciente índice (probandos), se puede considerar un diagnóstico prenatal.
Para prevenir el daño ocular permanente, es necesario implementar una inmunosupresión sistémica a largo plazo. Las opciones terapéuticas incluyen:
- corticosteroides sistémicos
- Metotrexato
- Ciclosporina
 Comprendiendo la Ciclosporina: Usos e Indicaciones Terapéuticas La ciclosporina es un fármaco inmunosupresor fundamental, empleado principalmente en el tratamiento de diversas enfermedades inflamatorias graves. Su aplicación se extiende a condiciones que afectan tanto la piel como otros órganos internos del cuerpo. En Nueva Zelanda, este tratamiento está accesible y completamente financiado en sus formulaciones de tabletas y líquido oral. Adicionalmente, existen versiones oftálmicas (gotas para los ojos) e inyectables, aunque más
Comprendiendo la Ciclosporina: Usos e Indicaciones Terapéuticas La ciclosporina es un fármaco inmunosupresor fundamental, empleado principalmente en el tratamiento de diversas enfermedades inflamatorias graves. Su aplicación se extiende a condiciones que afectan tanto la piel como otros órganos internos del cuerpo. En Nueva Zelanda, este tratamiento está accesible y completamente financiado en sus formulaciones de tabletas y líquido oral. Adicionalmente, existen versiones oftálmicas (gotas para los ojos) e inyectables, aunque más - Micofenolato de mofetiloMicofenolato de Mofetilo: Usos e Implicaciones Terapéuticas El micofenolato de mofetilo representa la formulación en sal del potente fármaco inmunosupresor conocido como ácido micofenólico. Esta estructura salina mejora significativamente la tolerancia del paciente y asegura una absorción corporal rápida y eficiente, tras la cual se metaboliza al compuesto activo, el ácido micofenólico. El mecanismo de acción principal del ácido micofenólico radica en su capacidad para inhibir la proliferación de linfocitos más
- TalidomidaTalidomida: Descifrando sus Usos Terapéuticos Actuales y Mecanismo de Acción A finales de la década de 1950, la talidomida irrumpió en el mercado como un medicamento revolucionario, prometiendo alivio contra el insomnio y las náuseas, especialmente en mujeres embarazadas. Trágicamente, su promesa se desvaneció rápidamente cuando se documentaron numerosos y graves defectos congénitos en los neonatos expuestos al fármaco in utero. Este descubrimiento llevó a su restricción mundial inmediata. Sin más
- Inhibidores del factor de necrosis tumor (como infliximab, etanercept o adalimumab)
- Antagonistas de la interleucina-1, como anakinraAnakinra: Agente Biológico para Enfermedades Reumatoides y Dermatológicas Anakinra es un agente biológico recombinante empleado primordialmente en el tratamiento de enfermedades reumáticas, como la artritis reumatoide. Recientemente, su utilidad se ha expandido, demostrando ser eficaz en el manejo de diversas afecciones cutáneas inflamatorias. Composición y Desarrollo de Anakinra Anakinra funciona como un antagonista del receptor de interleucina-1 (IL-1Ra), generado mediante tecnología de ADN recombinante, utilizando un sistema de expresión bacteriano más
La inflamación ocular es a menudo la manifestación más difícil de controlar y constituye la principal fuente de complicaciones crónicas.


