Was ist Waardenburg síndrome?
Das Waardenburg-Syndrom ist eine seltene genetischen Störung, die durch Taubheit und Anomalien Pigmentierung, wie zum Beispiel:
- Weiße Haarbüschel
- Hypopigmentierte Flecken (blasse) Haut
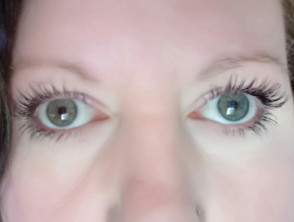
- Iris-Heterochromie (verschiedenfarbige Augen)
Das Waardenburg-Syndrom kann auch mit muskuloskelettaler Defekten und dem Hirschsprung-Syndrom in Verbindung gebracht werden.
Das Waardenburg-Syndrom ist benannt nach Petrus Johannes Waardenburg, einem niederländischen Ophthalmologen, der feststellte, dass Iris-Heterochromie häufig mit Taubheit einherging.
Waardenburg-Syndrom
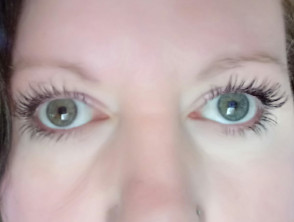
Iris-Heterochromie
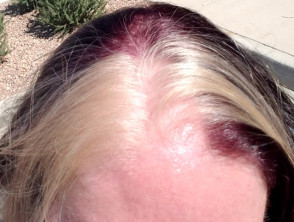
Weiße Haarbüschel
Wer bekommt das Waardenburg-Syndrom?
Das Waardenburg-Syndrom kann vererbt werden oder spontan auftreten. Es betrifft Männer und Frauen gleichermaßen und kann alle Rassen betreffen.
Es hat eine Populationshäufigkeit von 1 zu 42.000 und ist für 1-3% aller Fälle von angeborener angeboren Taubheit verantwortlich.
Was verursacht das Waardenburg-Syndrom?
Das Waardenburg-Syndrom wird als Neurokristopathie angesehen. Es ist das Ergebnis von Anomalien nicht abgeschlossen haben Kamm Differenzierung während der embryonalen Entwicklung.
Verschiedene Gen Mutationen kann das Waardenburg-Syndrom verursachen, was zu einigen Unterschieden in den Anzeichen und Symptomen führt. Expression und Penetranz sind ebenfalls variabel.
- WS1 und WS3 werden durch Mutationen im PAX3-Gen auf Chromosoms 2q
- WS2A - MITF (Mikrophthalmie-assoziierter Transkription Faktor)-Gen, Chromosom 3p
- WS2B - Chromosom 1p
- WS2C - Chromosom 8p
- WS2D: SNAI2-Gen, Chromosom 8q
- WS2E - SOX10-Gen, Chromosom 22q
- WS4A - EDNRB-Gen auf 13q
- WS4B - EDN3-Gen auf 20q
- WS4C - SOX10-Gen (wie bei WS2E)
Verschiedene Arten von Mutation können das Waardenburg-Syndrom verursachen: Insertionen, Deletionen, Frameshift-Mutationen, Spleißstellenanomalien, Nonsense- oder Stopp-Mutationen.
Die meisten Formen des Waardenburg-Syndroms sind autosomal-dominant dominant, was bedeutet, dass nur ein betroffenes Gen an ein Kind weitergegeben werden muss, damit es das Syndrom hat. Die Vererbung von Defekten in EDN3 oder EDNRB ist komplexer. Normalerweise sind sie autosomal-rezessiv, obwohl Fälle von autosomal autosomal-dominanten Vererbung dominanter Vererbung mit unvollständiger Penetranz beschrieben wurden.
Andere Mutationen in einigen der oben genannten Genen können verwandte klinische Syndrome verursachen, wie Tietz-Syndrom (MITF-Gen), Piebaldismus (SNAI2-Gen), PCWH (SOX10-Gen) oder ABCD-Syndrom (EDNRB-Gen).
Was sind die klinischen Merkmale des Waardenburg-Syndroms?
Die Merkmale sind von Geburt an vorhanden. Da es sich um eine seltene Erkrankung handelt und die klinischen Anzeichen subtil sein können, wird die Diagnose möglicherweise erst später im Leben gestellt.
Es wurden vier Haupt-Subtypen des Waardenburg-Syndroms identifiziert.
Typ 1
Typ 1 ist der häufigste Subtyp des Waardenburg-Syndroms. Zu den Merkmalen gehören:
- Neurosensorische Taubheit
- Typische Gesichtsstruktur mit Distopia canthorum (seitlicheseitliche Verschiebung der inneren Augenwinkel, breite Nasenwurzel und Synophrys (Zusammenwachsen der Augenbrauen in der Mittellinie) Mittellinie)
- Hypopigmentierte Flecken von Haut und Haare
- Pigmentstörungen Auffälligkeiten der Augen, wie Iris-Heterochromie (verschiedenfarbige Augen), die vollständig, teilweise oder segmentär sein kann; Isohypochromie der Iris (blassblaue Augen); oder Pigmentstörungen des Augenhintergrunds segmentalen; oder Pigmentstörungen des Augenhintergrunds Augenhintergrunds
Typ 2
Typ 2 weist ähnliche klinische Merkmale wie das Waardenburg-Syndrom Typ 1 auf, jedoch sind die inneren Augenwinkel normal.
Typ 3
Typ 3 (Klein-Waardenburg-Syndrom) weist ebenfalls ähnliche Merkmale wie das Waardenburg-Syndrom Typ 1 auf, jedoch mit muskuloskelettalen Anomalien wie Muskelhypoplasie, Beugenbereichen Gelenkkontrakturen o Syndaktylie (fusionierte Finger/Zehen).
Typ 4
Typ 4 (Shah-Waardenburg-Syndrom) weist ähnliche Merkmale wie das Waardenburg-Syndrom Typ 2 auf, jedoch mit dem Hirschsprung-Syndrom (einer Erkrankung, die durch das Fehlen von Nervus Nervenzellen in den Muskeln eines Teils oder des gesamten Dickdarms entsteht).
Wie wird das Waardenburg-Syndrom diagnostiziert?
Die Diagnose des Waardenburg-Syndroms basiert auf klinischen Merkmalen. Im Jahr 1992 entwickelte das Waardenburg-Konsortium Haupt- und Nebenkriterien für die Diagnose.
Hauptkriterien
- Neurosensorische Taubheit
- Pigmentstörung der Iris keine, wie Iris-Heterochromie: vollständig, teilweise oder segmentär; Isohypochromie der Iris; oder Pigmentstörungen des Augenhintergrunds
- Anomalien der Haar-Pigmentierung, wie weiße Haarbüschel, Augenbrauen oder Wimpern
- Distopia canthorum: seitliche Verschiebung des inneren Augenwinkels
- Erstgradiger Verwandter mit Waardenburg-Syndrom
Nebenkriterien
- Hypopigmentierte Hautflecken
- Synophrys
- Breite Nasenwurzel
- Hypoplasie der Nasenflügel
- Vorzeitige Alterung der Kopfhaut
Eine klinische Diagnose des Waardenburg-Syndroms Typ 1 erfordert 2 Hauptkriterien oder 1 Haupt- und 2 Nebenkriterien.
Der W-Index
Der W-Index kann berechnet werden, um festzustellen, ob eine Distopia canthorum vorliegt.
- a = Abstand der inneren Augenwinkel
- b = Pupillenabstand
- c = Abstand der äußeren Augenwinkel
- X = (2a- (0,2119c + 3,909)) / c
- Y = (2a- (0,2479b + 3,909)) / b
- W = X + Y + a / b
- Ein W-Index > 1,95 ist ein Hinweis auf eine Distopia canthorum.
Die genetische Sequenzierung des PAX3-Gens auf Mutationen, die das Waardenburg-Syndrom verursachen, kann als Teil der genetischen Beratung für Familienmitglieder durchgeführt werden. Pränatale Pränatale oder Chorionzottenbiopsie o Amniozentese, aber dies wird selten getan. Dies liegt an der klinischen Variabilität innerhalb des Waardenburg-Syndroms, da das Vorhandensein der Mutation nicht angibt, welche klinischen Merkmale vorhanden sein werden oder wie schwerwiegend diese sind.
Was ist die Behandlung für das Waardenburg-Syndrom?
Es gibt keine direkte Behandlung für das Waardenburg-Syndrom selbst, und da es sich um eine genetische Erkrankung handelt, gibt es keine Heilung. Genetische Beratung kann für betroffene Patienten, die eine Familie gründen möchten, von Vorteil sein.
- Bei Kindern mit Verdacht auf Waardenburg-Syndrom sollten audiologische Untersuchungen durchgeführt werden, um die Taubheit zu beurteilen. Hörgeräte oder Cochlea-Implantate können erforderlich sein.
- Patienten mit Hirschsprung-Syndrom benötigen möglicherweise eine Operation, um das betroffene Darmsegment zu entfernen.
- Die hypopigmentierten Hautflecken sind anfälliger anfällig für Sonnenschäden, daher ist Sonnenschutz wichtig.
Was ist die Prognose des Waardenburg-Syndroms?
Das Waardenburg-Syndrom ist eine Pruritus chronische Erkrankung und die Merkmale bleiben lebenslang bestehen. Die Lebenserwartung ist normal.


