Wat is Sturge – Weber syndroom?
Het Sturge-Weber-syndroom is zeldzaam, aangeborenen niet geërfd neurocutaan stoornis gekenmerkt door capillair misvorming op de huid van het gezicht (wijnvlek) en haarvatenveneuze misvormingen in de hersenen en ogen [1].
Sturge-Weber-syndroom
Sturge-Weber-syndroom
Wie krijgt het Sturge-Weber-syndroom?
Er wordt geschat dat ongeveer één op de 20.000 tot 50.000 baby's wordt geboren met het Sturge-Weber-syndroom [2]. Omdat het wordt veroorzaakt door een somatisch mozaïek mutatie, wordt niet geërfd van de ouders.
Wat veroorzaakt het Sturge-Weber-syndroom?
Het Sturge-Weber-syndroom wordt veroorzaakt door een somatische mozaïekmutatie van de GNAQ gen in chromosoom 9 [3]. Dit betekent dat de mutatie in het gen heeft plaatsgevonden in de lichaamscellen na de vorming van de zygoot. GNAQ reguleert intracellulair signaalwegen. De mutatie resulteert in de ongecontroleerde vorming of rijping van haarvaten in de aangetaste cellen.
Wat zijn de klinische kenmerken van het Sturge-Weber-syndroom?
Het Sturge-Weber-syndroom wordt gekenmerkt door vasculair misvormingen in het gezicht, de ogen en de hersenen van de getroffen mensen. Deze zijn bij de geboorte aanwezig.
- Wijnvlekken zijn de meest voorkomende vorm van vasculaire misvormingen en treffen ongeveer drie op de duizend baby's, maar de meeste zijn niet geassocieerd met het Sturge-Weber-syndroom. [4].
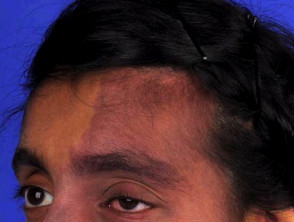
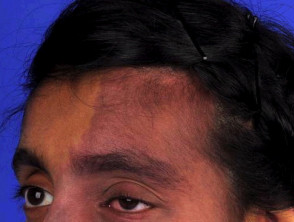
- Portwijnvlekken bij het Sturge-Weber-syndroom worden meestal aangetroffen in de verdeling van de eerste en tweede divisie van de trigeminus zenuw op het voorhoofd en het bovenste ooglid [5]. Ze kunnen ook beide zijden van het gezicht aantasten. [6].
- Leptomeningeale vasculaire malformaties ontstaan in de hersenen aan dezelfde kant als de wijnvlek.
- Leptomeningeale vasculaire misvormingen kunnen ook optreden zonder portwijnvlek [7].
Neurologisch Y oogheelkundig Tekenen van het Sturge-Weber-syndroom zijn dat wel progressief en meestal ontwikkelen in de eerste twee levensjaren. Deze kunnen het volgende omvatten:
- toevallen en epilepsie
- Hemiparese en beroerte-achtige gebeurtenissen
- gedragsproblemen
- Gezichtsvelddefecten en glaucoom
- Groeihormoondeficiëntie.
Wat zijn de complicaties van het Sturge-Weber-syndroom?
Complicaties van het Sturge-Weber-syndroom zijn afhankelijk van de omvang van vasculaire misvormingen en andere klinische kenmerken. Ze zijn uiterst variabel.
Hoe wordt het Sturge-Weber-syndroom gediagnosticeerd?
De diagnose van het Sturge-Weber-syndroom is gebaseerd op de vondst van de karakteristieke trigeminale wijnvlekken en leptomeningeale capillaire-veneuze malformaties. Een diagnose gebaseerd op leptomeningen verwondingen het hangt gewoon af van de ontwikkelen van symptomen.
Als er neurologische symptomen of bevindingen zijn, magnetische resonantie beeldvorming (Magnetische resonantie) van de hersenen wordt uitgevoerd met gadoliniumcontrast om capillair-veneuze leptomeningeale misvormingen op te sporen.
Welke is de differentiële diagnose voor het Sturge-Weber-syndroom?
De kenmerken van andere vasculaire malformatiesyndromen worden hieronder beschreven.
- Het Klippel-Trénaunay-syndroom heeft uitgebreidere capillaire misvormingen, treft de ledematen en de romp en gaat gepaard met hypertrofie van het aangedane ledemaat.
- Het Parkes-Weber-syndroom presenteert zich met een grote capillaire misvorming in één ledemaat en hypertrofie van de aangedane ledemaat. Er zijn meerdere snelstromende arterioveneuze shunts.
- Het Servelle-Martorell-syndroom, een zeldzame aangeboren angiodysplastische ziekte, wordt geassocieerd met progressieve hypotrofie van de ledematen.
- Het Proteus-syndroom presenteert zich met asymmetrisch en onevenredige overgroei van lichaamsdelen. Het syndroom is het resultaat van een somatisch activerende mutatie in de AKT1 oncogen.
- Het CLOVES-syndroom presenteert zich met congenitale lipomateuze overgroei, vasculaire malformatie, epidermaal naevus, wervelkolom/skelet/afwijkingenscoliose. Het syndroom is het gevolg van de activering van het somatische mozaïek mutaties in de PIK3CA gen.
Wat is de behandeling voor het Sturge-Weber-syndroom?
Er bestaat geen specifieke behandeling voor het Sturge-Weber-syndroom. De behandeling bestaat uit het beheersen van de huid, neurologische en oculair symptomen, met beperkt succes.
Behandeling van aanvallen bij patiënten met het Sturge-Weber-syndroom met anti-epileptica is niet altijd succesvol [8]. Er lijkt geen enkele behandeling te bestaan hoger aan anderen.
Portwijnvlekken kunnen worden behandeld met gepulseerde kleurstof Zijn met variabele resultaten [9]. Vanwege de over het algemeen slechte resultaten van gepulseerde kleurstof lasers alleen, huidig Er worden anti-angiogene middelen getest gehecht therapieën.
Omdat glaucoom een veel voorkomende complicatie is van het Sturge-Weber-syndroom, zijn regelmatige oogcontroles met een oogarts worden aanbevolen. Glaucoom wordt operatief en medisch behandeld [10].
Baby's waarbij het Sturge-Weber-syndroom is vastgesteld, moeten worden behandeld met een lage dosis aspirine [11,12]. Antitrombotische therapie kan de progressie van de ziekte voorkomen, wat de bloedtoevoer naar de hersenen kan beïnvloeden en tot neuronale schade kan leiden.
Wat is het resultaat van het Sturge-Weber-syndroom?
de voorspelling van het Sturge-Weber-syndroom hangt af van de mate van betrokkenheid van de hersenen en de huid. Uitgebreide portwijnvlekken worden in verband gebracht met een verhoogd risico op epilepsie en glaucoom bilateraal Leptomeningeale vasculaire malformaties worden geassocieerd met intellectuele en leerstoornissen. [13]. Het begin van aanvallen vóór de leeftijd van één jaar heeft een significant effect op de cognitieve en motorische functie bij kinderen met het Sturge-Weber-syndroom [14].
Er wordt geschat dat ongeveer één op de twee volwassenen met het Sturge-Weber-syndroom neurologische defecten heeft, zelfs bij degenen die er aanvankelijk last van hadden. asymptomatisch.