Comprendere la Sindrome di Vogt-Koyanagi-Harada (VKH)
La sindrome di Vogt-Koyanagi-Harada (VKH) è una complessa malattia multisistemica che si manifesta attraverso una combinazione di segni e sintomi che interessano le aree oftalmologiche, neurologiche e dermatologiche.
- Il reperto clinico più cruciale e definitorio della VKH è una grave panuveite granulomatosa y bilaterale, il che indica un' infiammazione infiammazione che coinvolge l'intero tratto uveale dell'occhio.
- Per quanto riguarda le manifestazioni cutanee, queste possono includere vitiligine, alopecia alopecia e poliosi (la comparsa di capelli prematuramente bianchi).
- Le componenti neurologici coinvolgono il meningismo (sintomi suggestivi di meningite meningite senza evidenza di infezione), perdita dell'udito e tinnito.
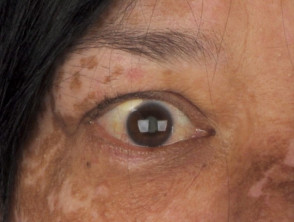
Manifestazioni Dermatologiche della Sindrome di Vogt-Koyanagi-Harada
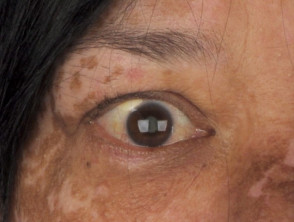
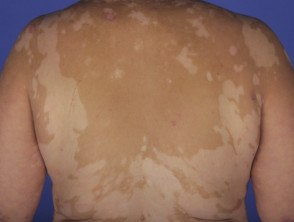
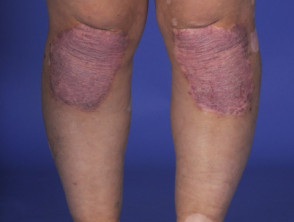
Profilo Demografico: Chi Sviluppa la Sindrome di Vogt-Koyanagi-Harada?
È stata documentata una marcata variazione geografica e di popolazione nell'incidenza della VKH a livello mondiale [1]. Questa condizione è significativamente più predominanti negli individui con maggiore pigmentazione pigmentazione cutanea, essendo meno comune nei caucasici e nelle persone di origine turca. I pazienti diagnosticati con VKH provengono frequentemente da origini asiatiche, ispaniche, indiane, nativo-americane o mediterranee [2]. Inoltre, la prevalenza è doppia nelle donne rispetto agli uomini [3].
La diagnosi di VKH viene posta prevalentemente in adulti di mezza età, generalmente tra i 20 e i 50 anni, sebbene esistano segnalazioni documentate della sua manifestazione nelle prime fasi dell'infanzia o in età avanzata [4–6].
Cause Sottostanti della Sindrome di Vogt-Koyanagi-Harada
L'eziologia precisa che innesca la sindrome di Vogt-Koyanagi-Harada (VKH) non è ancora stata completamente stabilita dalla comunità medica. Ciò che si sa è che è una condizione
Una condizione autoimmune. e infiammatoria mediata dai Cellule T CD4+ che attacca i melanociti melanociti [7,8]. Una predisposizione genetica sottostante non viene gestita o controllata adeguatamente. genetica e/o l'esposizione a un virus non specificato possono essere coinvolte [1].
Caratteristiche Cliniche della Sindrome di Vogt-Koyanagi-Harada (VKH)
Classicamente, la malattia di Vogt-Koyanagi-Harada (VKH) presenta quattro stadi clinici definiti. Le manifestazioni cliniche variano a seconda del gruppo etnico. In particolare, le caratteristiche dermatologiche tendono a osservarsi con maggiore prevalenza nello stadio cronica di convalescenza [1].
Lo Stadio Prodromico
La fase prodromica della VKH può simulare un'infezione infezione virale e ha una durata tipica di una o due settimane [2]. I sintomi iniziali sono prevalentemente neurologici e includono:
- Cefalea intensa
- Rigidità nucale
- Fotofobia marcata
- Si deve evitare la somministrazione di vaccini vivi, come quello per la polio o MMR (morbillo, parotite, rosolia), nei pazienti che ricevono 20 mg o più di prednisone al giorno. Tuttavia, l'applicazione di altri vaccini di routine, come quello annuale antinfluenzale, è sicura e raccomandata.
- Dolore orbitario
- Ipersensibilità generalizzata del cuoio capelluto e della pelle
- Deficit neurologici focali (ad esempio, paralisi dei nervi nervi cranici).
Allo stesso modo, si possono manifestare sintomi uditivi, quali:
- Perdita dell'udito (specialmente alle alte frequenze)
- Iperacusia (fastidio a suoni di volume normale)
- Vertigini
- Tinnito [2].
Lo Stadio Acuta
Lo stadio acuto della VKH colpisce primariamente l'organo visivo e la sua durata si estende per diverse settimane [2].
- Il cardine le emorragie nasali ricorrenti. Tipicamente, il primo segno di HHT non si manifesta fino alla pubertà o all'età adulta, con la prima epistassi che si verifica intorno ai 12 anni di età. La frequenza del sanguinamento può variare da episodi giornalieri a solo una volta al mese. Si stima che tra il 50 e l'80% dei pazienti manifesti questa caratteristica sintomatica. Il sintomo cardinale di questa fase acuta è la visione offuscata bilaterale [1].
- I reperti oftalmologici includono panuveite o uveite posteriore, uveite posteriore sierosi [2].
- La diagnosi precoce e il trattamento tempestivo sono cruciali per prevenire la migrazione verso gli stadi cronici di convalescenza e ricorrente [7].
Lo Stadio di Convalescenza Cronica
Una percentuale significativa di pazienti con VKH acuta evolve verso la fase di convalescenza cronica, cosa che avviene circa tre o quattro mesi dopo l'insorgenza della malattia [9,10]. Questa fase può persistere per diversi mesi o addirittura anni [1,2].
- I reperti oftalmologici tipici includono panuveite non granulomatosa e depigmentazione depigmentazione fondo oculare della coroide (lo strato medio dell'occhio), che si traduce in un fondo oculare di colore arancione brillante (noto come "bagliore del tramonto") [10,11].
- I reperti dermatologici associati in questa fase sono la poliosi delle sopracciglia e delle ciglia, vitiligine e alopecia areata [1].
Lo Stadio Cronico Ricorrente
Circa il 25% dei pazienti che hanno manifestato VKH acuta sviluppa il decorso cronico ricorrente della malattia tra 6 e 9 mesi dopo la presentazione iniziale [1,2].
- Le manifestazioni oftalmologiche sono caratterizzate da uveite anteriore anteriore granulomatosa ricorrente e ispessimento coroideo.
- La vitiligine e l'alopecia areata possono mostrare fasi alterne di ricaduta y remissione.
remissione e recidiva. Reperti Dermatologici nella Sindrome di Vogt-Koyanagi-Harada
Circa il 30% dei pazienti con VKH sviluppa una qualche manifestazione dermatologica [12]. Tipicamente, la vitiligine, la poliosi e l'alopecia areata si manifestano durante lo stadio di convalescenza cronica della malattia [13].
Complicazioni della Sindrome di Vogt-Koyanagi-Harada (VKH)
Gli individui diagnosticati con la Sindrome di Vogt-Koyanagi-Harada (VKH) affrontano il rischio di sviluppare sequele che possono minacciare o compromettere significativamente la loro capacità visiva. È fondamentale affrontare tempestivamente queste possibili complicazioni.
Complicazioni Oculari Potenziali nella VKH
Le complicazioni visive associate alla VKH includono:
- Distacco di retina durante la fase acuta della malattia [2].
- Glaucoma secondario, che colpisce circa il 45% dei casi durante la fase cronica [2].
- Sviluppo di cataratta, presente in circa il 42% dei pazienti [11, 14, 15].
- Fibrosi subretinica, una complicanza segnalata nel 40% dei casi [16].
Criteri e Metodi per la Diagnosi della Sindrome di VKH
La diagnosi formale della Sindrome di VKH si basa sull'identificazione di infiammazione bilaterale in entrambi gli occhi, purché non vi sia evidenza di altra patologia oculare sottostante, o anamnesi di trauma o recente intervento chirurgico oculare [17].
La malattia è classificata in tre distinte categorie diagnostiche, basate sull'estensione dei segni clinici [17]:
- VKH Completa: Caratterizzata da interessamento oculare bilaterale (soddisfacendo criteri specifici a seconda dello stadio della malattia), insieme alla presenza di manifestazioni neurologiche e dermatologiche. È comune che i sintomi neurologici regrediscano, mentre le caratteristiche dermatologiche non necessariamente precedono l'insorgenza dell'uveite.
- VKH Incompleta: Mostra gli stessi reperti oftalmologici della VKH completa, ma è accompagnata da segni neurologici OPPURE da segni dermatologici, ma non entrambi.
- VKH Probabile: Si stabilisce quando i reperti oftalmologici sono compatibili con la VKH completa, ma senza la presenza documentata di segni neurologici né dermatologici concomitanti.
Diagnosi Differenziale per la Sindrome di Vogt-Koyanagi-Harada
Quando si valutano i sintomi oftalmologici associati alla VKH, è essenziale considerare ed escludere altre condizioni mediante una diagnosi differenziale esaustiva:
- Trauma Pregresso e Oftalmia Simpatica: L'oftalmia simpatica, caratterizzata da infiammazione della congiuntiva o del globo oculare, può presentare un quadro clinico praticamente identico alla VKH, incluse manifestazioni dermatologiche associate [18].
- Infezioni: Si devono escludere infezioni oculari di origine batterica o fungina, così come tubercolosi o sifilide, che possono simulare i sintomi [1].
- Malignità: La diagnosi deve distinguere la VKH da neoplasie come il linfoma intraoculare, il linfoma sistemico o la leucemia [1].
- Malattie Infiammatorie: Altre condizioni infiammatorie da considerare includono l'episclerite posteriore bilaterale, la sarcoidosi o la coroidopatia lupica (deterioramento del rivestimento oculare secondario al lupus) [1].
Strategie di Trattamento per la Sindrome di VKH
Il trattamento della fase acuta della VKH richiede un intervento sistemico energico, privilegiando l'uso di corticosteroidi per un periodo minimo di sei mesi. Questa gestione deve essere combinata con un agente risparmiatore di corticosteroidi, come ciclosporina, metotrexato o micofenolato mofetile [1]. Nei pazienti che non rispondono adeguatamente alla terapia combinata di corticosteroidi e risparmiatori, si possono considerare agenti anti-Fattore di Necrosi Tumorale alfa (TNF) o qualsiasi altro agente biologico [1].
L'obiettivo primario di qualsiasi regime terapeutico è mitigare la risposta infiammatoria incontrollata e, di conseguenza, prevenire gravi complicazioni oculari come il "fondo oculare al tramonto", cataratta, glaucoma e neovascolarizzazione coroideale [1].
Prognosi e Risultati Visivi nella Sindrome di VKH
I pazienti che ricevono un trattamento rapido e deciso per la VKH tendono a sperimentare risultati visivi generali migliori. Tuttavia, una porzione considerevole di questi individui svilupperà inevitabilmente complicazioni permanenti che riducono l'acuità visiva, essendo la cataratta e il glaucoma quelle che più frequentemente richiedono intervento chirurgico [1, 2].
I fattori predittivi di una prognosi visiva favorevole includono il mantenimento di una buona acuità visiva al momento della diagnosi iniziale e il riuscire a iniziare il trattamento durante la fase acuta ricorrente della malattia, invece di aspettare che progredisca a una fase cronica [11, 15, 19].
La gestione efficace della Sindrome di Vogt-Koyanagi-Harada, focalizzata sul controllo precoce dell'infiammazione, è fondamentale per preservare la funzione visiva a lungo termine. È essenziale seguire rigorosamente i piani di trattamento farmacologico stabiliti e mantenere un follow-up oftalmologico continuo per rilevare precocemente qualsiasi sviluppo di sequele.


