Entendiendo el Síndrome de Vogt-Koyanagi-Harada (VKH)
El síndrome de Vogt-Koyanagi-Harada (VKH) es una compleja enfermedad multisistema que se manifiesta a través de una combinación de signos y síntomas que afectan las áreas oftalmológicas, neurológicas y dermatológicas.
- El hallazgo clínico más crucial y definitorio del VKH es una grave panuveítis granulomatosa y bilateral, lo que indica una inflamación que abarca todo el tracto uveal del ojo.
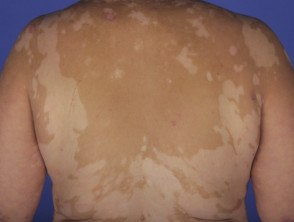
- En cuanto a las manifestaciones cutáneas, estas pueden incluir vitíligo, alopecia areata y poliosis
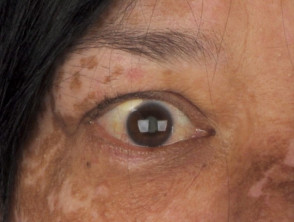
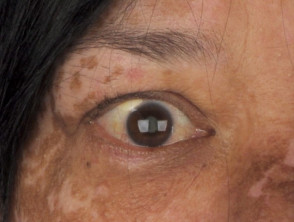
 Entendiendo la Poliosis: Causas y Condiciones Asociadas La poliosis se define como la aparición de un parche localizado de cabello blanco. Esta despigmentación resulta de la ausencia de melanina, el pigmento encargado de dar color, en los tallos capilares de la zona afectada. Aunque es más visible en el cuero cabelludo, puede manifestarse también en cejas, pestañas u otras áreas pilosas del cuerpo. El desarrollo de la poliosis puede deberse más (la aparición de cabello prematuramente blanco).
Entendiendo la Poliosis: Causas y Condiciones Asociadas La poliosis se define como la aparición de un parche localizado de cabello blanco. Esta despigmentación resulta de la ausencia de melanina, el pigmento encargado de dar color, en los tallos capilares de la zona afectada. Aunque es más visible en el cuero cabelludo, puede manifestarse también en cejas, pestañas u otras áreas pilosas del cuerpo. El desarrollo de la poliosis puede deberse más (la aparición de cabello prematuramente blanco). - Los componentes neurológicos abarcan el meningismo (síntomas sugestivos de meningitis sin evidencia de infección), pérdida auditiva y tinnitus.
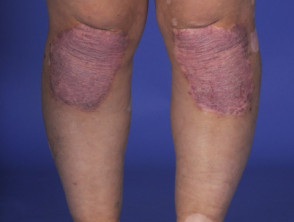
Manifestaciones Dermatológicas del Síndrome de Vogt-Koyanagi-Harada
Perfil Demográfico: ¿Quién Desarrolla el Síndrome de Vogt-Koyanagi-Harada?
Se ha documentado una marcada variación geográfica y poblacional en la incidencia del VKH a nivel mundial [1]. Esta afección es significativamente más predominante en individuos con mayor pigmentación de la piel, siendo menos común en caucásicos y personas de ascendencia turca. Los pacientes diagnosticados con VKH frecuentemente provienen de orígenes asiáticos, hispanos, indios, nativos americanos o mediterráneos [2]. Adicionalmente, la prevalencia es el doble en mujeres que en hombres [3].
El diagnóstico del VKH se realiza predominantemente en adultos de mediana edad, generalmente entre los 20 y 50 años, aunque existen reportes documentados de su manifestación en etapas tempranas de la infancia o en la vejez [4–6].
Causas Subyacentes del Síndrome de Vogt-Koyanagi-Harada
La etiología precisa que desencadena el síndrome de Vogt-Koyanagi-Harada (VKH) todavía no ha sido completamente establecida por la comunidad médica. Lo que sí se sabe es que es una condición
Una afección autoinmune e inflamatoria mediada por Células T CD4+ que ataca a los melanocitos [7,8]. Una predisposición genética y/o la exposición a un virus no especificado pueden estar involucradas [1].
Características Clínicas del Síndrome de Vogt-Koyanagi-Harada (VKH)
Clásicamente, la enfermedad de Vogt-Koyanagi-Harada (VKH) presenta cuatro estadios clínicos definidos. Las manifestaciones clínicas varían según el grupo étnico. En particular, las características dermatológicas tienden a observarse con mayor prevalencia en la etapa crónica de convalecencia [1].
La Etapa Prodrómica
La fase prodrómica del VKH puede simular una infección viral y tiene una duración típica de una a dos semanas [2]. Los síntomas iniciales son predominantemente neurológicos e incluyen:
- Cefalea intensa
- Rigidez cervical
- Fotofobia marcada
- Fiebre
- Dolor orbitario
- Hipersensibilidad generalizada del cuero cabelludo y la piel
- Déficits neurológicos focales (por ejemplo, parálisis de nervios craneales).
Asimismo, se pueden manifestar síntomas auditivos, tales como:
- Pérdida auditiva (especialmente en las frecuencias altas)
- Hiperacusia (molestia ante sonidos de volumen normal)
- Vértigo
- Tinnitus [2].
La Etapa Aguda
La etapa aguda de VKH afecta primariamente al órgano visual y su duración se extiende por varias semanas [2].
- El síntoma cardinal de esta fase aguda es la visión borrosa bilateral [1].
- Los hallazgos oftalmológicos incluyen panuveítis o uveítis posterior, acompañados de desprendimientos de retina multifocales y serosos [2].
- La identificación temprana y el tratamiento oportuno son cruciales para prevenir la migración hacia las etapas crónicas de convalecencia y recurrente [7].
La Etapa de Convalecencia Crónica
Una proporción significativa de pacientes con VKH aguda evoluciona a la fase de convalecencia crónica, lo cual ocurre aproximadamente tres a cuatro meses después del inicio de la enfermedad [9,10]. Esta fase puede persistir durante varios meses o incluso años [1,2].
- Los hallazgos oftalmológicos típicos incluyen panuveítis no granulomatosa y despigmentación de la coroides (la capa media del ojo), lo que resulta en un fondo de ojo de color naranja brillante (conocido como "resplandor del atardecer") [10,11].
- Los hallazgos dermatológicos asociados en esta etapa son la poliosis de cejas y pestañas, vitiligo y alopecia areata
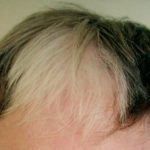
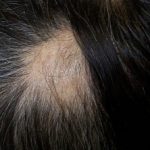 Comprendiendo la Alopecia Areata: Definición y Factores de Riesgo El término alopecia hace referencia a la pérdida anormal del cabello (alopecia). Específicamente, la alopecia areata se manifiesta con la aparición súbita de uno o más parches circulares de calvicie, localizados con mayor frecuencia en el cuero cabelludo. A esta condición también se le conoce como alopecia autoinmune, y en ciertas presentaciones puede manifestarse como pérdida capilar difusa. Imágenes Ilustrativas de más [1].
Comprendiendo la Alopecia Areata: Definición y Factores de Riesgo El término alopecia hace referencia a la pérdida anormal del cabello (alopecia). Específicamente, la alopecia areata se manifiesta con la aparición súbita de uno o más parches circulares de calvicie, localizados con mayor frecuencia en el cuero cabelludo. A esta condición también se le conoce como alopecia autoinmune, y en ciertas presentaciones puede manifestarse como pérdida capilar difusa. Imágenes Ilustrativas de más [1].
La Etapa Crónica Recurrente
Aproximadamente el 25% de los pacientes que experimentaron VKH aguda desarrollan el curso crónico recurrente de la enfermedad entre 6 y 9 meses después de la presentación inicial [1,2].
- Las manifestaciones oftalmológicas se caracterizan por uveítis anterior granulomatosa recurrente y engrosamiento coroideo.
- El vitiligo y la alopecia areata pueden exhibir fases alternantes de recaída y remisión.
Hallazgos Dermatológicos en el Síndrome de Vogt-Koyanagi-Harada
Alrededor del 30% de los pacientes con VKH desarrollan alguna manifestación dermatológica [12]. Típicamente, el vitiligo, la poliosis y la alopecia areata se manifiestan durante la etapa de convalecencia crónica de la enfermedad [13].
Complicaciones del Síndrome de Vogt-Koyanagi-Harada (VKH)
Los individuos diagnosticados con el Síndrome de Vogt-Koyanagi-Harada (VKH) enfrentan el riesgo de desarrollar secuelas que pueden amenazar o deteriorar significativamente su capacidad visual. Es crucial abordar estas posibles complicaciones con prontitud.
Complicaciones Oculares Potenciales en VKH
Las complicaciones visionarias asociadas al VKH incluyen:
- Desprendimiento de retina durante la fase aguda de la enfermedad [2].
- Glaucoma secundario, que afecta aproximadamente al 45% de los casos durante la fase crónica [2].
- Desarrollo de cataratas, presente en cerca del 42% de los pacientes [11, 14, 15].
- Fibrosis subretiniana, una complicación reportada en el 40% de los casos [16].
Criterios y Métodos para el Diagnóstico del Síndrome de VKH
El diagnóstico formal del Síndrome de VKH se basa en la identificación de inflamación bilateral en ambos ojos, siempre y cuando no exista evidencia de otra patología ocular subyacente, o antecedentes de trauma o cirugía ocular reciente [17].
La enfermedad se clasifica en tres categorías diagnósticas distintas, basándose en la extensión de los signos clínicos [17]:
- VKH Completo: Caracterizado por afectación ocular bilateral (cumpliendo criterios específicos según el estadio de la enfermedad), junto con la presencia de manifestaciones neurológicas y dermatológicas. Es común que los síntomas neurológicos remitan, mientras que las características dermatológicas no preceden necesariamente al inicio de la uveítis.
- VKH Incompleto: Muestra los mismos hallazgos oftalmológicos que el VKH completo, pero se acompaña de signos neurológicos O bien, signos dermatológicos, pero no ambos.
- VKH Probable: Se establece cuando los hallazgos oftalmológicos son compatibles con el VKH completo, pero sin la presencia documentada de signos neurológicos ni dermatológicos acompañantes.
Diagnóstico Diferencial para el Síndrome de Vogt-Koyanagi-Harada
Cuando se evalúan los síntomas oftalmológicos asociados a VKH, es esencial considerar y descartar otras afecciones mediante un diagnóstico diferencial exhaustivo:
- Traumatismo Previo y Oftalmía Simpática: La oftalmía simpática, caracterizada por inflamación de la conjuntiva o el globo ocular, puede presentar un cuadro clínico prácticamente idéntico al VKH, incluyendo manifestaciones dermatológicas asociadas [18].
- Infecciones: Se deben descartar infecciones oculares de origen bacteriano o fúngico, así como la tuberculosis o la sífilis, que pueden simular los síntomas [1].
- Malignidad: El diagnóstico debe distinguir el VKH de neoplasias como el linfomaComprendiendo el Linfoma: Definición y Clasificaciones El linfoma se define como una maligna proliferación anormal de linfocitos. La categorización de los distintos tipos de linfomas es inherentemente compleja y su clasificación está en constante evolución, impulsada por los avances en la comprensión de su origen y comportamiento biológico [1–3]. El proceso diagnóstico del linfoma habitualmente requiere la obtención de una muestra mediante biopsia de un linfa nodo, tejido circundante o más intraocular, el linfoma sistémico o la leucemia [1].
- Enfermedades Inflamatorias: Otras condiciones inflamatorias a considerar incluyen la escleritis posterior bilateral, la sarcoidosis
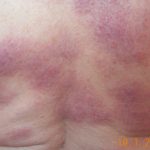 Sarcoidosis: Definición, Riesgos y Enfermedades Desencadenantes La sarcoidosis se define como un trastorno inflamatorio multisistémico, caracterizado por la formación de granulomas. Estos son cúmulos localizados de células inflamatorias que pueden afectar diversas partes del cuerpo. Desde una perspectiva patológica, estos granulomas se describen típicamente como epitelioides no caseificantes, lo que los diferencia claramente de los granulomas caseificantes asociados a dolencias como la tuberculosis. Usualmente, esta condición se origina en los más o la coroidopatía lúpica (deterioro del revestimiento ocular secundario al lupus) [1].
Sarcoidosis: Definición, Riesgos y Enfermedades Desencadenantes La sarcoidosis se define como un trastorno inflamatorio multisistémico, caracterizado por la formación de granulomas. Estos son cúmulos localizados de células inflamatorias que pueden afectar diversas partes del cuerpo. Desde una perspectiva patológica, estos granulomas se describen típicamente como epitelioides no caseificantes, lo que los diferencia claramente de los granulomas caseificantes asociados a dolencias como la tuberculosis. Usualmente, esta condición se origina en los más o la coroidopatía lúpica (deterioro del revestimiento ocular secundario al lupus) [1].
Estrategias de Tratamiento para el Síndrome de VKH
El tratamiento de la fase aguda del VKH requiere una intervención sistémica enérgica, priorizando el uso de corticoesteroides durante un periodo mínimo de seis meses. Este manejo debe combinarse con un agente ahorrador de corticosteroides, tales como ciclosporina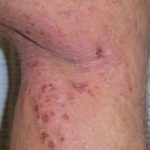 Comprendiendo la Ciclosporina: Usos e Indicaciones Terapéuticas La ciclosporina es un fármaco inmunosupresor fundamental, empleado principalmente en el tratamiento de diversas enfermedades inflamatorias graves. Su aplicación se extiende a condiciones que afectan tanto la piel como otros órganos internos del cuerpo. En Nueva Zelanda, este tratamiento está accesible y completamente financiado en sus formulaciones de tabletas y líquido oral. Adicionalmente, existen versiones oftálmicas (gotas para los ojos) e inyectables, aunque más, metotrexato o micofenolato de mofetiloMicofenolato de Mofetilo: Usos e Implicaciones Terapéuticas El micofenolato de mofetilo representa la formulación en sal del potente fármaco inmunosupresor conocido como ácido micofenólico. Esta estructura salina mejora significativamente la tolerancia del paciente y asegura una absorción corporal rápida y eficiente, tras la cual se metaboliza al compuesto activo, el ácido micofenólico. El mecanismo de acción principal del ácido micofenólico radica en su capacidad para inhibir la proliferación de linfocitos más [1]. En pacientes que no responden adecuadamente a la terapia combinada de corticosteroides y ahorradores, se pueden considerar agentes anti-Factor de Necrosis Tumoral alfa (TNF) o cualquier otro agente biológico [1].
Comprendiendo la Ciclosporina: Usos e Indicaciones Terapéuticas La ciclosporina es un fármaco inmunosupresor fundamental, empleado principalmente en el tratamiento de diversas enfermedades inflamatorias graves. Su aplicación se extiende a condiciones que afectan tanto la piel como otros órganos internos del cuerpo. En Nueva Zelanda, este tratamiento está accesible y completamente financiado en sus formulaciones de tabletas y líquido oral. Adicionalmente, existen versiones oftálmicas (gotas para los ojos) e inyectables, aunque más, metotrexato o micofenolato de mofetiloMicofenolato de Mofetilo: Usos e Implicaciones Terapéuticas El micofenolato de mofetilo representa la formulación en sal del potente fármaco inmunosupresor conocido como ácido micofenólico. Esta estructura salina mejora significativamente la tolerancia del paciente y asegura una absorción corporal rápida y eficiente, tras la cual se metaboliza al compuesto activo, el ácido micofenólico. El mecanismo de acción principal del ácido micofenólico radica en su capacidad para inhibir la proliferación de linfocitos más [1]. En pacientes que no responden adecuadamente a la terapia combinada de corticosteroides y ahorradores, se pueden considerar agentes anti-Factor de Necrosis Tumoral alfa (TNF) o cualquier otro agente biológico [1].
El objetivo primordial de cualquier régimen terapéutico es mitigar la respuesta inflamatoria descontrolada y, consecuentemente, prevenir complicaciones oculares graves como el "fondo de ojo al atardecer", cataratas, glaucoma y la neovascularización coroidea [1].
Pronóstico y Resultados Visuales en el Síndrome de VKH
Los pacientes que reciben un tratamiento rápido y decidido para el VKH suelen experimentar mejores resultados visuales generales. No obstante, una porción considerable de estos individuos inevitablemente desarrollará complicaciones permanentes que restan agudeza visual, siendo las cataratas y el glaucoma las que con mayor frecuencia exigen intervención quirúrgica [1, 2].
Los factores predictivos de un pronóstico visual favorable incluyen mantener una buena agudeza visual al momento del diagnóstico inicial y lograr iniciar el tratamiento durante la etapa aguda recurrente de la enfermedad, en lugar de esperar a que progrese a una fase crónica [11, 15, 19].
La gestión efectiva del Síndrome de Vogt-Koyanagi-Harada, enfocada en el control temprano de la inflamación, es fundamental para preservar la función visual a largo plazo. Es esencial seguir rigurosamente los planes de tratamiento farmacológico establecidos y mantener un seguimiento oftalmológico continuo para detectar tempranamente cualquier desarrollo de secuelas.


