Ictiosis Arlequín: Abordaje Clínico Integral
introduzione
La ictiosis arlequín es una de las genodermatosis más raras y graves observadas en la práctica dermatológica y neonatológica. Su reconocimiento temprano es fundamental, ya que constituye una urgencia médica que requiere manejo multidisciplinario intensivo. En las últimas décadas, los avances en cuidados neonatales, terapias de soporte y retinoides sistémicos han mejorado significativamente la supervivencia y calidad de vida de los pacientes.
Definición y características generales
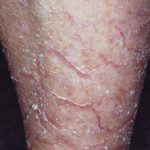
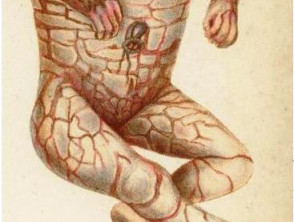
La ictiosis arlequín es una forma severa de ictiosis congénita autosómica recesiva caracterizada por la formación de placas gruesas de hiperqueratosis que envuelven al recién nacido como una “armadura”. Estas placas se acompañan de profundas fisuras eritematosas y alteraciones anatómicas faciales y acrales.
Es importante no confundirla con otras condiciones que llevan el término “arlequín”, tales como:
- Síndrome de arlequín: una alteración autonómica que provoca pérdida segmentaria de sudoración y cambios unilaterales de rubor.
- Cambio de color arlequín en neonatos: fenómeno transitorio de eritema unilateral.
- Feto Arlecchino: denominación histórica para la forma congénita severa de esta ictiosis.
Fisiopatologia
La causa primaria es una mutación en el gen ABCA12, responsable de codificar un transportador de lípidos esencial en la formación de la barrera epidérmica.
- Las mutaciones severas provocan pérdida total de función, impidiendo el transporte adecuado de lípidos hacia el estrato córneo.
- La ausencia de estos lípidos altera la cohesión celular y genera disfunción profunda de la barrera cutánea, lo que explica:
- hiperqueratosis masiva,
- fisuras dolorosas,
- incapacidad para regular temperatura y fluidos,
- alta susceptibilidad a infecciones.
Epidemiología y factores de riesgo
- Incidencia aproximada: 1 en 300.000 nacidos vivos.
- Afecta por igual a ambos sexos.
- No presenta predisposición étnica.
- La incidencia puede aumentar en poblaciones con consanguineità.
Caratteristiche cliniche
Manifestazioni cutanee
- Presentes desde el nacimiento.
- Placas gruesas, brillantes y rígidas de ipercheratosi.
- Fisuras profundas y eritematosas entre placas.
- Movilidad corporal severamente limitada.
Compromiso facial
- Ectropión severo, con exposición de conjuntiva y riesgo elevado de queratitis e infecciones.
- Hipoplasia nasal, narinas obstruidas y erosión de las alas nasales.
- Orejas aplanadas, poco desarrolladas o ausentes; el conducto auditivo puede estar obturado por escamas.
- Eclabium (eversión labial), que dificulta la alimentación y favorece deshidratación.
Estremità
- Piel rígida y gruesa que limita la movilidad.
- Puede haber hipoplasia digital, sindactilia o incluso polidactilia.
- Riesgo de constricción y autoamputación por presión de la piel hiperqueratósica.
Termorregulación y función de barrera
- Sudoración reducida o ausente → riesgo de hipertermia.
- Pérdida elevada de fluidos → deshidratación.
- Compromiso respiratorio por restricción torácica.
Complicazioni
- Infecciones cutáneas y sistémicas potencialmente mortales.
- Contracturas y constricción vascular de extremidades, con riesgo de necrosis.
- Insufficienza respiratoria por rigidez de la pared torácica.
- Alteraciones oculares graves por exposición.
- Dificultad para alimentarse y mantener el balance hidroelectrolítico.
Diagnosi
Diagnóstico clínico
- Características cutáneas típicas presentes desde el nacimiento.
- Alteraciones faciales y acrales altamente sugestivas.
Estudios complementarios
- Confirmación genética mediante detección de mutaciones en ABCA12: prueba de mayor especificidad diagnóstica.
- Biopsia cutánea: hiperqueratosis masiva y alteraciones epidérmicas, útil cuando el diagnóstico genético no está disponible.
- Mutaciones menos severas pueden producir:
- membrana de colodión,
- eritrodermia ictiosiforme congénita.
Diagnosi differenziale
Incluye otros tipos de ictiosis congénitas y síndromes asociados:
- Ittiosi volgare (mutación en FLG).
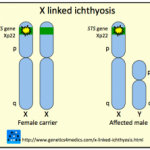
- Ictiosis recesiva ligada al X (deficiencia de esteroide sulfatasa).
- Ictiosis autosómica recesiva congénita (lamelar, eritrodermia ictiosiforme).
- Ictiosis queratinopáticas (mutaciones en genes de queratina).
- Sindrome di Sjögren-Larsson: ictiosis + parálisis cerebral + deterioro intelectual.
- Enfermedad de Gaucher neonatal: puede presentarse con fenotipo de “bebé colodión”.
- Tricotiodistrofia: fotosensibilidad, cabello frágil, retraso en el crecimiento.
- Condrodisplasia punctata ligada al X (Conradi-Hünermann-Happle): ictiosis en parches, alopecia cicatricial, cataratas, malformaciones óseas.
Trattamento
Manejo inicial en neonatología
La ictiosis arlequín constituye una emergencia neonatal que requiere cuidados intensivos:
- Aislamiento térmico y control estricto del ambiente.
- Hidratación adecuada y manejo de electrolitos.
- Profilaxis y tratamiento precoz de infecciones.
- Humidificación constante para evitar sequedad.
- Lubricación ocular intensiva por riesgo de daño corneal.
Cuidado cutáneo
- Emolientes de alta potencia aplicados frecuente y cuidadosamente.
- Especialmente útiles los que contienen urea, acido salicilico o alfa-hidroxiácidos, siempre bajo supervisión médica por la vulnerabilidad cutánea.
- Mantener la piel flexible disminuye fisuras y reduce riesgo de infección.
Retinoides sistémicos
- El uso temprano de acitretina o isotretinoína ha demostrado:
- acelerar la separación de placas,
- suavizar la piel,
- mejorar la movilidad,
- aumentar la supervivencia.
- Deben ser manejados por especialistas debido a riesgos y efectos secundarios.
Pronóstico y seguimiento
Históricamente, la mortalidad neonatal era extremadamente alta. Con el manejo moderno, las tasas de supervivencia global se acercan al 50%.
Los sobrevivientes pueden evolucionar hacia un fenotipo de ictiosis menos severo, pero suelen presentar:
- Escamas persistentes tipo “pez”.
- Acúmulo seborreico de material ceroso.
- Alopecia cicatricial y pobre crecimiento del cabello.
- Alteraciones en termorregulación.
- Contracturas digitales y artralgias.
- Hipotiroidismo y baja talla.
- Necesidad de cuidados dermatológicos durante toda la vida.
El seguimiento debe ser multidisciplinare: dermatología, neonatología, nutrición, oftalmología, genética y kinesiología.
Prevención y asesoramiento genético
- No existe una prevención primaria específica.
- El asesoramiento genético es fundamental en familias afectadas.
- La detección de portadores y el diagnóstico prenatal mediante estudio de ABCA12 son opciones disponibles en centros especializados.
conclusione
La ictiosis arlequín es una genodermatosis devastadora que requiere diagnóstico inmediato y manejo intensivo y coordinado. Los avances en fisiopatología, cuidados neonatales y uso temprano de retinoides sistémicos han transformado el pronóstico de una enfermedad antes considerada casi invariablemente fatal. El cuidado continuado permite a muchos pacientes alcanzar la infancia y vida adulta, aunque con importantes desafíos dermatológicos y sistémicos. Un enfoque integral y basado en la evidencia es clave para mejorar su supervivencia y calidad de vida.
Feto Arlecchino
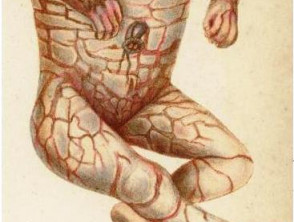
Feto Arlecchino
Relazionato: