Cos'è il virus di Epstein-Barr?
Il virus di Epstein-Barr (EBV), noto anche come herpesvirus umano 4 (HHV-4), è uno degli otto herpesvirus linfotropici conosciuti [1].
- EBV il più delle volte causa la mononucleosi infettiva, nota anche come ghiandolare febbre, caratterizzato da febbre, mal di gola e linfoadenopatia [2].
- La maggior parte delle persone è esposta a EBV durante i primi decenni di vita e lo è asintomatico [23]. Dopo il primario infezione, EBV rimane latente In memoria Cellule B.e per la maggior parte delle persone, non ha gravi conseguenze sulla salute. [4].
- In alcuni casi, EBV può potenziare (aumentare l'efficienza della) trasformazione dei linfociti B e causare a linfoproliferativa disturbo.
Qual è Patogenesi di infezione da EBV?
La patogenesi di EBV si adatta al seguente processo [2].
- EBV infetta principalmente il epitelio del orofaringe (parte posteriore della gola) e salivare ghiandole ed è sparso da queste cellule.
- Il virus infetta quindi i linfociti B vicini, direttamente nelle tonsille. cripte (tasche o pieghe) o dopo il contatto con il epiteliale cellule.
- Le cellule B infette circolanti si diffondono quindi attraverso il flusso sanguigno.
- il proliferazione dei linfociti B infettati da EBV porta all'allargamento linfoide tessuti, compresi linfa ghiandole.
- I linfociti B infettano qualsiasi reagente Cellule T. con cui entrano in contatto.
- Il virus si trasmette da persona a persona attraverso le secrezioni orali.
Quali sono i disordini linfoproliferativi associati a EBV?
I disturbi linfoproliferativi associati a EBV sono rari; i cui criteri sono:
- Uno o più tipi di cellule linfoidi infettate da EBV
- Le cellule linfoidi infette possono dividersi eccessivamente, portando a sviluppo di una benigno disturbo o a malignità [5]
- I tumori maligni associati a EBV includono i linfociti B linfoma, Cellula T. linfomi e tumori non linfoproliferativi, rinofaringeo e gastrico carcinoma [2].
Di conseguenza possono verificarsi disturbi linfoproliferativi associati a EBV latenza deregolamentazione con:
- Virus dell'immunodeficienza umana (HIV) infezione e immunodeficienza acquisita sindrome (AIDS)
- Altre forme di immunodeficienza
- Immunosoppressore terapia dopo solido Organo trapianto
- Senescenza immunitaria legata all'età (deterioramento del sistema immunitario dovuto all'età) [4,6].
Come vengono classificati i disturbi linfoproliferativi da EBV?
La classificazione dei disordini linfoproliferativi associati a EBV si basa sul lignaggio delle cellule bersaglio, delle cellule B, T e delle cellule natural killer (NK). [7].
I disordini linfoproliferativi a cellule B associati a EBV includono [7]:
- Linfoma di Burkitt
- Linfoma di Hodgkin classico
- Spedire-trapianto disturbo linfoproliferativo
- Disturbi linfoproliferativi associati all'HIV
- Disturbi linfoproliferativi correlati ad altri istotipi (tipi di tessuto dalla crescita di a tumore) [7].
I disturbi linfoproliferativi a cellule NK e T associati a EBV includono [7]:
- Periferica Linfoma a cellule T.
- Cronico infezione attiva da EBV dei tipi di cellule T e NK (con cutaneo e sistemico forme)
- Linfoma angioimmunoblastico a cellule T
- Extranodal Linfoma nasale a cellule T/NK.
EBV è stato implicato più direttamente negli ultimi due tipi [7].
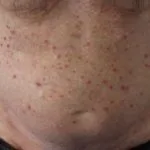
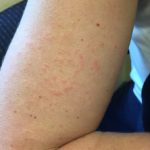
Cosa sono la pelle dimostrazioni di disordini linfoproliferativi associati a VPD?
Le caratteristiche cutanee dei disordini linfoproliferativi associati a EBV derivano principalmente da cellule T infette o Cellule NK. Queste manifestazioni cutanee si osservano in:
- Linfoma extranodale a cellule NK/T [1]
- EBV positivo mucocutanea ulcera (EBV-MCU) [5]
- Linfomatoide granulomatosi
- Linfoma plasmablastico (PBL) [8].
Linfoma extranodale a cellule NK/T
Secondo l'attuale classificazione dell'Organizzazione Mondiale della Sanità (OMS) dei tumori ematolinfoidi, NK extranodale /Cellula T. Il linfoma è una forma rara ma estremamente aggressiva di linfoma. [7]. La maggior parte dei pazienti con questo tipo di linfoma presenta cellulite o ulcera facciale. [8].
Le sottocategorie si basano sui siti anatomici di coinvolgimento. [8]:
- Il linfoma "nasale" a cellule NK/T di solito colpisce il tratto digestivo superiore
- Il linfoma a cellule NK/T di "tipo nasale" colpisce la pelle, Tessuto morbido, tratto gastrointestinale e testicoli.
Quando il linfoma extranodale a cellule NK/T si presenta inizialmente con segni cutanei, è noto come linfoma cutaneo primitivo extranodale a cellule NK/T. Il linfoma nasale a cellule NK/T può anche presentarsi con metastasi [9].
Il linfoma extranodale a cellule NK/T di tipo nasale di solito include nodulie meno spesso ulcerazione nell'addome e nelle estremità [10]. Il linfoma a cellule NK/T di tipo nasale è clinicamente meno aggressivo e più trova rispetto al tipo nasale.
La diagnosi di linfoma extranodale a cellule NK/T dipende dalla sua stadiazione e comporta il completamento delle seguenti indagini [9]:
- Nasello flessibileendoscopia con biopsie per valutare il coinvolgimento nasale
- Midollo osseo biopsia
- Connecticut esame del torace, dell'addome e del bacino
- Biopsia cutanea di qualsiasi pelle sospetta. lesione.
Istologicamente, il linfoma extranodale a cellule NK/T è caratterizzato da EBV positivo atipico linfoide citotossico infiltrarsi, vascolare distruzione e tessuto necrosi (vedi pagina DermNet NZ sul linfoma extranodale a cellule NK/T, tipo nasale, patologia).
Il trattamento del linfoma extranodale a cellule NK/T è dipendente nella sua messa in scena [9].
- Sistemico chemioterapia di solito viene offerto.
- Radioterapia è utile nei pazienti con malattia di stadio I o II limitata alla cavità nasale.
- Antivirale La terapia viene utilizzata per i pazienti con un carico di EBV elevato.
- Può essere preso in considerazione il trapianto allogenico di cellule staminali ematopoietiche.
- Monoclonale anticorpi sono oggetto di indagine.
Ulcera mucocutanea positiva al virus di Epstein-Barr
EBV-MCU è stato aggiunto alla classificazione dell'OMS 2016 ed è una condizione localizzata che non coinvolge il linfonodi, midollo osseo, fegato o milza. Tende a presentarsi come un solitario doloroso, behdemarcato ulcera nell'orofaringe, nella pelle o nel tratto gastrointestinale e può essere associata a perdita di peso [5].
I fattori predisponenti per EBV-MCU includono:
- Trattamento con immunosoppressori [11]:
- Metotrexato
- Azatioprina
- Tacrolimus
- Micofenolato
- Fattore di necrosi tumorale alfa inibitori
- attuale steroidi
- immunosenescenza legata all'età
- Immunodeficienze primarie [3].
La patogenesi di EBV-MCU correla con una diminuzione della popolazione di cellule T in immunosoppressi pazienti, con conseguente proliferazione di ristretti cloni di cellule T specifiche per EBV nel corpo. Ciò porta a una linfoproliferazione localizzata guidata da EBV, poiché il sistema immunitario può solo mantenere dormiente il virus. [undici].
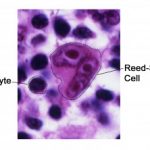
La diagnosi lavoro per EBV-MCU di solito implica istologico valutazione con immunoistochimica. La carica virale EBV è generalmente negativa. Istologicamente, il lesioni sono costituiti da infiltrati di linfociti, plasma cellule, istiocitie eosinofili, con grandi esplosioni di cellule B atipiche che ricordano le cellule di Hodgkin Reed-Sternberg. Come risultato delle loro caratteristiche istologiche simili ad altre cellule B proliferativo tumori, diagnosi errata del linfoma di Hodgkin classico o diffondere grande Cella B si è verificato un linfoma [6].
Il decorso della malattia da EBV-MCU in genere aumenta e diminuisce ed è relativamente benigno. I pazienti possono spontaneamente fare riferimento o avere una risposta clinica completa a terapie immunosoppressive ridotte. Tuttavia, alcuni casi hanno persistente decorso debilitante che richiede una terapia aggressiva [3,11].
Le opzioni di trattamento includono:
- Anticorpi monoclonali come quelli diretti al CD20 anticorpo terapia (p. es., rituximab) o terapia con anticorpi diretti contro il CD30 (p. es., brentuximab)
- radioterapia locale
- chirurgico locale escissione
- chemioterapia sistemica
- Terapia combinata.
Granulomatosi linfomatoide
La granulomatosi linfomatoide è una malattia rara in cui vi è una sovrapproduzione di cellule B anormali infettate da EBV. [12]. Le cellule si infiltrano e si accumulano in vari tessuti del corpo.
I sintomi della granulomatosi linfomatoide variano a seconda degli organi colpiti.
- La granulomatosi linfomatoide dei polmoni provoca mancanza di respiro, tosse e dolore toracico.
- Altri siti interessati dalla granulomatosi linfomatoide includono il sistema nervoso centrale, i reni, il fegato e la pelle.
- Possono verificarsi sintomi sistemici, come malessere, febbre e perdita di peso.
La granulomatosi linfomatoide cutanea può presentarsi con [12]:
- Macule o patch
- Papule o piatti
- Sottocutaneo o dermico noduli
- Ulcerazione.
La biopsia della granulomatosi linfomatoide tende a mostrare un infiltrato di cellule B atipiche e polimorfo Cellule T, con infiammazione e necrotica focolai all'interno delle cellule linfoidi [13]. Una biopsia non è sempre affidabile, poiché potrebbero mancare le caratteristiche cellule anormali.
Il trattamento della granulomatosi linfomatoide dipende dal numero di cellule B EBV-positive e dall'entità della necrosi. [12]. Alcuni pazienti possono regredire spontaneamente, ma la maggior parte richiede un trattamento con interferone alfa-2b o chemioterapia di combinazione con rituximab.
linfoma plasmablastico
PBL è un sottotipo raro ma aggressivo di linfoma diffuso a grandi cellule B [14]. È spesso associato all'immunosoppressione sottostante nei pazienti con HIV o trapianto di organi solidi, ma la PBL può colpire anche i pazienti immunocompetenti. [quindici]. Molti casi di PBL sono associati a EBV, il che è importante terapeutico trascendenza [14]. Alcuni studi hanno riportato che la positività all'EBV non prevede l'esito, mentre altri hanno riportato che denota meglio previsione [16].
Il PBL tende ad essere limitato alla cavità orale e alla mascella, ma può anche interessare la pelle, i linfonodi, il midollo osseo, i polmoni e l'intestino. [17]. Nell'ambiente post-trapianto, la pelle e i linfonodi sono i siti più comuni di malattia. distribuzione [18].
Il PBL cutaneo primario è molto raro. Le caratteristiche cliniche includono noduli viola, eritematoso placche infiltrate e ulcerative infiltrato lesioni alle gambe [15,17].
Il trattamento per PBL non è stato standardizzato. Le opzioni di trattamento includono:
- Chemioterapia
- Escissione chirurgica
- Trattamento combinato
- Terapia antiretrovirale altamente attiva per PBL correlato all'HIV.
La prognosi di PBL è sfavorevole, con mediano Sopravvivenza a 8 mesi [16].