Wat is familie Middellandse Zee koorts?
Familiaire mediterrane koorts is erfelijk auto-inflammatoir syndroom gekarakteriseerd door terugkerend korte periodes van hoge koorts geassocieerd met buikpijn, ontsteking van gewrichten en andere delen van het lichaam en de huid uitslag. Indien niet behandeld, amyloïdose het ontwikkelt zich gewoonlijk en kan dodelijk zijn. Familiale mediterrane koorts is de meest voorkomende van de periodieke koortssyndromen.
Bij familiale mediterrane koorts type 2 is amyloïdose het presenterende kenmerk zonder voorgeschiedenis koorts afleveringen.
Wie krijgt familiale mediterrane koorts en waarom?
Familiaire mediterrane koorts treft vooral specifieke raciale groepen die afkomstig zijn uit het oostelijke Middellandse Zeegebied. Deze omvatten:
- Arabieren
- Armeniërs
- Italianen (milde vorm)
- Joden
- Turken.
In deze groepen wordt 1 op de 25 tot 1 op de 2.000 mensen getroffen. Ter vergelijking, in West-Europeanen, de overwicht het is 2,5 per 100.000 (1 op 40.000) mensen. de vervoerder het percentage kan in sommige groepen oplopen tot 1 op de 3. Hoewel voorheen werd aangenomen dat het alleen Sefardische joden treft, is onlangs erkend dat Asjkenazische joden lijden aan een milde vorm van het syndroom. Tweederde van de patiënten ontwikkelen symptomen vóór 5 jaar en 90% na 20 jaar.
Familiaire mediterrane koorts wordt meestal geërfd als een autosomaal recessieve aandoening, wat betekent dat beide ouders de afwijking moeten dragen gen en er is een kans van één op vier dat een kind wordt getroffen (MIM 249100). Er is een minder gebruikelijke autosomaal dominant formulier (MIM 134610).
moleculair biologie en genetica
Familiale mediterrane koorts is meestal te wijten aan mutaties in de mediterrane koortsMEFV) of pyrine-gen gelokaliseerd in chromosoom 16 (16p13.3). mutaties in de MEFV gen is gevonden in de 80% van typische gevallen. Er zijn meer dan 100 verschillende ziektegerelateerde mutaties geïdentificeerd. De meest gerapporteerde mutatie, M694V, wordt geassocieerd met ernstige ziekte en ontwikkelen van secundaire amyloïdose. Andere veel voorkomende mutaties zijn M680I en V726A. 20% van Asjkenazische joden draagt de E148Q MEFV mutatie.
de MEFV genetische codes voor een eiwit genaamd pyrin (ook bekend als marenostrine) voornamelijk uitgedrukt door opruiend witte bloedcellen zoals neutrofielen Y eosinofielen. De normale vorm van pyrine onderdrukt ontstekingen door pro-inflammatoire stoffen naar beneden te reguleren. cytokinen (mobiele boodschapper eiwitten), opwaartse regeling van ontstekingsremmend cytokines en voorkomen de productie van interleukine 1beta (IL-1β) door ontstekingsmaskers. Het ontbreken van functionele pyrine maakt de inflammatoire cascade vrij.
Er zijn ook milieueffecten op de demonstraties van familiale mediterrane koorts. Armeniërs met familiale mediterrane koorts die in Armenië wonen, hebben bijvoorbeeld een incidentie van amyloïdose varieert van 25 tot 48%, terwijl Armeniërs die in de VS wonen een incidentie hebben van amyloïdose van 0%.
Wat zijn de klinische kenmerken van familiale mediterrane koorts?
De meest voorkomende klinische kenmerken van familiaire mediterrane koorts zijn:
- koortsaanvallen die 1 tot 3 dagen duren
- ernstige buik-, borst- en/of gewrichtspijn
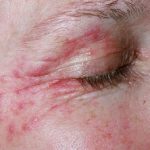
- erysipelas-achtige veranderingen op de onderbenen.
Het begin is bijna altijd vóór de leeftijd van 30 jaar. Kinderen jonger dan 2 jaar presenteren zich vaak alleen met koorts en Voortgang tot meer typische aanvallen op de leeftijd van 5 jaar.
De kenmerken van de terugkerende scherp Episodes van familiale mediterrane koorts worden beschreven in de onderstaande tabel.
| Symptoom | Functies |
|---|---|
| Koorts |
|
| Buikpijn |
|
| Pijn op de borst |
|
| Gewrichtspijn en zwelling (artralgie, artritis) |
|
| Huid huiduitslag |
|
| pijn in het scrotum (orchitis) |
|
| Vergrote milt (splenomegalie) |
|
| Spierpijn (spierpijn) |
|
| Neurologisch symptoom |
|
| amyloïdose |
|
Wat zijn de kenmerken van de huiduitslag?
| Vergelijkbaar met erysipelas verwondingen |
|
| Henoch-Schönlein Purper |
|
| niet-specifieke purpura |
|
| Polyarteritis nodosa |
|
| angio-oedeem |
|
| Roodheid van de handpalmen |
|
| Het fenomeen van Raynaud |
|
Wat triggert de aanvallen?
De frequentie van de afleveringen is variabel, van een week tot meerdere jaren. Aanvallen kunnen worden veroorzaakt door:
- Oefening
- Infectie
- Menstruatie
- Spanning.
Acute aanvallen lossen spontaan op. Tussen afleveringen is de gezondheid normaal.
Hoe wordt familiale mediterrane koorts gediagnosticeerd?
De diagnose familiale mediterrane koorts wordt gesteld aan de hand van de Tel-Hashomer-criteria:
- De diagnose is definitief als aan 2 major criteria of 1 major + 2 minor criteria is voldaan.
- De diagnose is waarschijnlijk als aan 1 major + 1 minor criteria is voldaan.
Belangrijkste criteria:
- Recidiverende febriele episodes geassocieerd met peritonitis, pleuritis of synovitis
- Type AA-amyloïdose zonder predisponerende ziekte
- Gunstige reactie op dagelijkse colchicine.
kleine criteria
- Terugkerende koortsepisodes
- Vergelijkbaar met erysipelas erytheem
- Positieve voorgeschiedenis van familiale mediterrane koorts bij een familielid in de eerste graad.
Onderzoek naar familiale mediterrane koorts
Bloedonderzoek tijdens een aanval kan het volgende laten zien:
- Verhoogd aantal witte bloedcellen (leukocytose), erytrocyt sedimentatiesnelheid (ESR), serum fibrinogeen, C-reactief eiwit (CRP)
- Verhoogd serum-IgD in een 10%.
Röntgenfoto's van de buik laten vaak meerdere vloeistofniveaus zien, wat duidt op een 'acute buik'
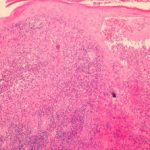
een huid biopsie van erysipelas blessure kan een zware tonen infiltreren van neutrofielen (witte bloedcellen). leukocytoclastisch vasculitis het is erin gezien biopsieën van Henoch-Schönlein purpura of laesies die lijken op polyarteritis nodosa.
Onderzoek naar myalgie (spierpijn) laat normale spieren zien enzymen, elektromyografie (EMG) en spierbiopsie.
Amyloïdose presenteert zich meestal als proteïnurie zonder rode bloedcellen of verhoogde bloeddruk.
Gensequencing kan worden uitgevoerd door gespecialiseerde laboratoria als gerichte mutatieanalyse (op zoek naar de meest voorkomende mutaties) of sequentieanalyse van geselecteerde exonen (kijkend naar exon 10 en mogelijk andere). Zodra de genetische mutaties bij de patiënt zijn vastgesteld, kan de dragerschap van de ouders, screening op andere bloedverwanten in de eerste graad, zelfs als asymptomatischY prenataal de diagnose kan worden gesteld. Identificatie van asymptomatische maar genetisch aangetaste individuen betekent dat een behandeling kan worden gestart om de ontwikkeling van amyloïdose te voorkomen.
Wat is de behandeling van familiale mediterrane koorts?
Colchicine, dat levenslang dagelijks oraal wordt ingenomen, is het favoriete medicijn bij familiale mediterrane koorts voor:
- verminder de frequentie van aanvallen
- de ernst van de aanvallen verminderen
- secundair voorkomen systemisch amyloïdose.
Colchicine is potentieel giftig Daarom is het erg belangrijk om geen overmatige dosis te nemen.
De dosis colchicine wordt geleidelijk verhoogd tot een maximale dosis van 2,5 mg/dag. De dosering wordt meestal bepaald door de frequentie en ernst van de aanvallen. Colchicine resulteert in een duidelijke verbetering van de symptomen bij 90-95%-patiënten en 75% heeft een kwijtschelding. Het is niet bekend hoe het werkt.
Andere behandeling
Acute aanvallen worden behandeld met steroïdeloze ontstekingsremmers (NSAID's) en pijnverlichting.
Langdurige koorts met spierpijn reageert op systemische koorts corticosteroïden.
De meest voorkomende reden voor het niet reageren op colchicine is een slechte therapietrouw als gevolg van gastro-intestinaal ongemak. 5-10% reageert echter niet op colchicine en dit kan hieraan te wijten zijn ABCB1 gen polymorfismen die de opname van colchicine door mononucleaire cellen beïnvloedt.
Thalidomide en biologische agentia, zoals dagelijks onderhuids anakinra (een interleukine-1 ontvanger antagonist) of etanercept, kunnen worden overwogen om aanvallen en de ontwikkeling van amyloïdose te voorkomen. Om de ernst van een aanval te verminderen, is gemeld dat een enkele dosis methylprednisolon of anakinra aan het begin van de episoden de symptomen verlicht.