Cos'è Wiskott-Aldrich? sindrome?
La sindrome di Wiskott-Aldrich (WAS) è una rara malattia da immunodeficienza ereditaria che causa infezioni ed è anche associato a microtrombocitopenia (bassa piastrina conta piastrinica e dimensioni ridotte in modo anomalo), eczema, un aumento del rischio di autoimmune malattie e alcuni tipi di Cancro.
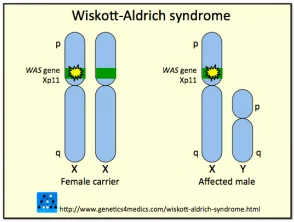
La sindrome è dovuta a mutazioni o eliminazioni in a gene trovato nella X cromosoma che codifica per la sindrome di Wiskott-Aldrich Proteina (VESPA). La proteina WAS è essenziale nella segnalazione e nell'organizzazione citoscheletrica (strutturale) delle cellule ematopoietiche (del sangue). Diversi tipi di mutazione all'interno del gene WASP può variare da individuo a individuo risultando in presenza dell'intero spettro clinico in alcuni casi, o solo alcune delle caratteristiche in altri.
Sono state descritte circa 160 diverse mutazioni o delezioni nel gene WASP. Poiché il gene che codifica per WASP è collegato al cromosoma X, la maggior parte dei casi con sindrome di Wiskott-Aldrich sono maschi; le ragazze possono essere raramente colpite. Si stima che la sindrome di Wiskott-Aldrich si manifesti in circa 1 a 10 su ogni milione di bambini. Le donne con il gene anormale generalmente non sono colpite vettori che trasmettono la mutazione alla generazione successiva.
Come viene classificata la sindrome di Wiskott-Aldrich?
La sindrome di Wiskott-Aldrich può essere considerata come uno spettro che presenta caratteristiche che rientrano tra le caratteristiche gravi della classica sindrome di Wiskott-Aldrich e la forma meno grave chiamata X-linked. trombocitopenia (XLT). I pazienti possono cambiare in gravità, a seconda della progressione della malattia nel tempo o della comparsa di complicanze come sviluppo di linfoma.
Quali sono i sintomi della sindrome di Wiskott-Aldrich?
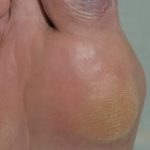
La trombocitopenia può essere presente dalla nascita e causare sanguinamento prolungato da ombelicale cavo. Anche la trombocitopenia causa petecchie (indicare il sanguinamento sulla pelle) e lividi (ecchimosi) che può verificarsi senza lesioni. La trombocitopenia può anche causare sanguinamento intestinale orale e nasale e intracranica sanguinamento Il sanguinamento può essere pericoloso per la vita.
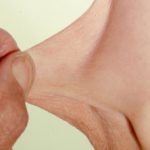
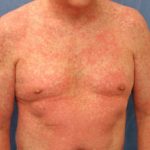
L'eczema colpisce l'80% dei pazienti con sindrome di Wisckott-Aldrich. L'eczema di solito compare durante l'infanzia o la prima infanzia. Le caratteristiche dell'eczema sono indistinguibili atopica eczema. I pazienti con sindrome di Wiskott-Aldrich hanno spesso livelli elevati di IgE e sviluppare allergie.
L'immunodeficienza può influenzare sia T che B Linfocita funzione. L'immunodeficienza aumenta il rischio e la frequenza di un'ampia gamma di infezioni, inclusa la secrezione dalle orecchie, batterica o polmonite virale, infezioni batteriche della pelle e herpes simplex (virus dell'herpes labiale). Polmone opportunistico infezione con Pneumocystis jiroveci può succedere.
La sindrome di Wiskott-Aldrich può essere associata a un'ampia gamma di malattie autoimmuni, più comunemente autoimmuni emolitico anemia, cutaneo vasculite, artrite e malattie renali. I pazienti affetti possono avere più malattie autoimmuni allo stesso tempo.
I tumori maligni più comuni associati alla sindrome di Wisckott-Aldrich sono leucemia e linfoma a cellule B.
Quali sono i risultati della pelle nella sindrome di Wiskott-Aldrich?
Ci sono una varietà di reperti cutanei nella sindrome di Wiskott-Aldrich:
- Cause della sindrome di Wiskott-Aldrich acuto e cronica eczema indistinguibile dall'eczema atopico. La sua gravità e persistenza sono variabili; nella sua forma più grave, è resistente a molti dei comuni trattamenti per l'eczema disponibili.
- Petecchie e lividi secondari al graffio eczematoso pelle o spontaneamente su pelle senza graffi.
- Infezioni opportunistiche della pelle come mollusco contagioso, herpes simplex e batteri. setticemia può anche essere persistente o ricorrente problemi per i pazienti con sindrome di Wiskott-Aldrich.
Genetica della sindrome di Wiskott-Aldrich *
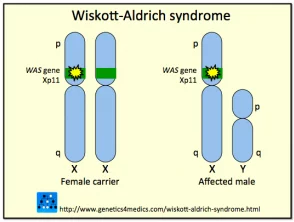
Sindrome di Wiskott Aldrich
* Immagine per gentile concessione di Genetics 4 Medics
Quali test possono essere utilizzati nella diagnosi della sindrome di Wiskott-Aldrich?
Nel test emocromocitometrico completo, la conta piastrinica è quasi sempre bassa e il piastrine sono caratteristicamente più piccoli del normale. In alcuni casi, la conta piastrinica rientra nell'intervallo normale, ma la dimensione delle piastrine è sempre influenzata. il neutrofili e anche la conta dei linfociti può essere bassa.
Se un neonato o un bambino con eczema presenta segni di trombocitopenia e sospetta immunodeficienza, deve essere eseguito un esame emocromocitometrico completo. Se nello striscio di sangue è presente trombocitopenia o piccole piastrine, si consiglia di rivolgersi a un pediatra.
Se c'è un forte sospetto clinico di sindrome di Wiskott-Aldrich e c'è un emocromo completo anomalie, il caso dovrebbe sempre essere discusso con a pediatrico immunologo prima di condurre ulteriori ricerche.
Immunoglobuline (anticorpo) i livelli nel flusso sanguigno possono essere bassi nella sindrome di Wiskott-Aldrich. La sindrome di Wiskott-Aldrich classica è associata a:
- bassi livelli di immunoglobulina M (IgM) e immunoglobulina G (IgG)
- I livelli di immunoglobulina A (IgA) e di immunoglobulina E (IgE) sono da normali ad alti.
Tuttavia, i bambini piccoli, in particolare, potrebbero non mostrare le classiche anomalie delle immunoglobuline perché la sindrome di Wiskott-Aldrich è associata a un graduale declino immunologico funzione.
Le risposte immunoglobuliniche alla vaccinazione possono essere assenti, specialmente le risposte ai vaccini pneumococcici.
Il test diagnostico di conferma per la sindrome di Wiskott-Aldrich è genetico analisi del sangue linfociti per identificare se esiste una specifica mutazione o delezione all'interno del gene WASP.
Quali sono le opzioni di trattamento per la sindrome di Wiskott-Aldrich?
Cellule staminali ematopoietiche trapianto, che di solito è un trapianto di midollo osseo, è curativo per i pazienti con sindrome di Wiskott-Aldrich. Per quelli con un fratello donatore geneticamente abbinato (circa 20% di pazienti), c'è un tasso di sopravvivenza di 80% e il tasso di sopravvivenza è solo leggermente inferiore per un trapianto di donatore abbinato non imparentato. Il tasso di sopravvivenza è inferiore e le complicanze sono più comuni per i trapianti da donatori correlati incompatibili. Il risultato è migliore se il trapianto di cellule staminali ematopoietiche viene eseguito nella prima infanzia.
precedente a trapianto o se il trapianto non è un'opzione, i bambini affetti potrebbero aver bisogno di un trattamento specifico per immunodeficienza e problemi di sanguinamento, tra cui:
- Trimetoprim-sulfametossazolo profilassi giornalmente per prevenire l'infezione da Pneumocystis jiroveci
- Profilassi con aciclovir per prevenire le infezioni da herpes simplex
- Infusioni periodiche di immunoglobuline per via endovenosa in cui sono presenti anomalie Cella B funzione
- Piastrine irradiate e trasfusioni di globuli rossi per il trattamento di episodi emorragici gravi.
I vaccini vivi come BCG e MMR lo sono controindicato. Anti-steroidi non steroideiinfiammatorio I farmaci (ibuprofene, diclofenac, aspirina e altri) dovrebbero essere evitati.
La terapia genica per la sindrome di Wiskott-Aldrich rimane sperimentale.
Outlook Qual è la prospettiva per i bambini con sindrome di Wiskott-Aldrich?
L'aspettativa di vita media dei bambini con sindrome di Wiskott-Aldrich è di circa 15-20 anni senza trapianto di cellule staminali ematopoietiche. I bambini trapiantati dovrebbero sopravvivere molto più a lungo.
Di quali informazioni genetiche hanno bisogno le famiglie?
Una volta genetica anomalia viene identificato nel bambino affetto, sua madre può essere testata per vedere se è portatrice del gene. Se lo fai, hai un rischio 50% di avere altri bambini affetti. Esiste anche il rischio 50% di trasmettere il gene alle figlie, che diventano portatrici e potrebbero averne affetti. Prenatale La diagnosi è disponibile per le famiglie colpite.