«`html
Entendiendo el Síndrome de Wiskott-Aldrich (WAS)
El síndrome de Wiskott-Aldrich (WAS) constituye un trastorno hereditario de inmunodeficienciaComprensión de la Inmunodeficiencia: Causas, Tipos y Consecuencias ¿Qué Provoca la Inmunodeficiencia y Cómo se Manifiesta? La inmunodeficiencia surge de un funcionamiento inadecuado del sistema inmunológico. Este estado puede ser desencadenado por una amplia variedad de factores, incluyendo síndromes hereditarios, infecciones, uso de ciertos medicamentos, condiciones médicas subyacentes, el embarazo, el proceso natural de envejecimiento y muchas otras causas. La gravedad de esta alteración es variable, lo que da lugar más poco frecuente. Se caracteriza por una susceptibilidad elevada a las infecciones, y a menudo se presenta junto con microtrombocitopenia (un recuento bajo de plaquetas y un tamaño plaquetario reducido anómalo), eczema notable, un riesgo incrementado de padecer enfermedades autoinmunes, y la aparición de ciertos tipos de cáncer.
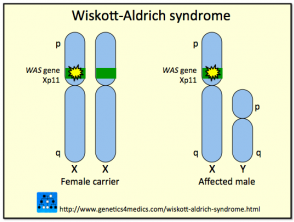
Este síndrome es el resultado de mutaciones o deleciones en un gen ubicado en el cromosoma X, el cual codifica la Proteína del Síndrome de Wiskott-Aldrich (WASP). La proteína WASP es crucial para la señalización celular y la organización citoesquelética (estructural) en las células hematopoyéticas (sanguíneas). La naturaleza y el tipo de mutación dentro del gen WASP pueden variar significativamente entre los afectados, lo que explica por qué algunos individuos manifiestan el espectro clínico completo, mientras que otros solo presentan algunas de las características.
Se han documentado cerca de 160 mutaciones o deleciones distintas en el gen WASP. Dado que este gen está ligado al cromosoma X, la gran mayoría de los casos diagnosticados con el síndrome de Wiskott-Aldrich corresponden a varones; las mujeres se ven afectadas con muy poca frecuencia. Se estima una prevalencia de 1 a 10 casos por cada millón de niños nacidos. Las mujeres portadoras del gen anómalo suelen permanecer asintomáticas, actuando como portadores que transmiten la mutación a la descendencia.
Clasificación y Espectro Clínico del Síndrome de Wiskott-Aldrich
El síndrome de Wiskott-Aldrich puede ser interpretado como un espectro de presentación clínica. Este varía desde las manifestaciones graves del WAS clásico hasta una forma menos severa conocida como trombocitopenia ligada al cromosoma X (XLT). La gravedad puede cambiar con el tiempo, influenciada por la progresión natural de la afección o por el desarrollo de complicaciones secundarias, como la linfomaComprendiendo el Linfoma: Definición y Clasificaciones El linfoma se define como una maligna proliferación anormal de linfocitos. La categorización de los distintos tipos de linfomas es inherentemente compleja y su clasificación está en constante evolución, impulsada por los avances en la comprensión de su origen y comportamiento biológico [1–3]. El proceso diagnóstico del linfoma habitualmente requiere la obtención de una muestra mediante biopsia de un linfa nodo, tejido circundante o más.
Manifestaciones Clínicas y Síntomas del WAS
La trombocitopenia puede ser evidente desde el momento del nacimiento, manifestándose como un sangrado prolongado del cable umbilical. Esta deficiencia plaquetaria también provoca la aparición de petequias (pequeños puntos hemorrágicos bajo la piel) y equimosis (hematomas) que pueden surgir sin un traumatismo previo. Adicionalmente, la trombocitopenia conlleva riesgos de hemorragias nasales, gastrointestinales y, en casos graves, hemorragias intracraneales. Este componente hemorrágico puede poner en peligro la vida del paciente.
El eccema afecta aproximadamente al 80% de las personas diagnosticadas con el síndrome de Wisckott-Aldrich, manifestándose Típicamente durante la infancia temprana. Las características dermatológicas del eccema en estos pacientes no se distinguen del eccema atópico común. Es frecuente que los pacientes con WAS presenten niveles elevados de Inmunoglobulina E (IgE) y, consecuentemente, desarrollar alergias múltiples.
La inmunodeficiencia asociada puede influir en la capacidad del cuerpo para combatir patógenos.
```
Tanto los linfocitos T como los B presentan una función alterada. La inmunodeficiencia incrementa la probabilidad y la frecuencia de diversas infecciones, incluyendo otitis media, neumonía bacteriana o viral, infecciones cutáneas bacterianas y el virus del herpesClasificación y Características de los Virus del Herpes Humano (HHV) Los virus del herpes humano (HHV) engloban diversos patógenos comunes que presentan manifestaciones cutáneas significativas. La familia Herpesvirales comparte varias características biológicas distintivas: • Poseen un genoma de ADN de doble cadena. • Incluyen una cápside de simetría icosaédrica, rodeada por un tegumento. • Están envueltos en una membrana lipídica, derivada de las membranas celulares infectadas pero modificada por la más simplex (VHS). Puede presentarse una infección pulmonar oportunista causada por *Pneumocystis jiroveci*.
El síndrome de Wiskott-Aldrich puede estar vinculado a un amplio espectro de trastornos autoinmunes, siendo los más comunes la anemia hemolítica autoinmune, la vasculitis cutánea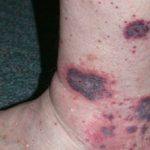 ¿Qué es la Vasculitis Cutánea? La vasculitis cutánea se define como un conjunto de afecciones caracterizadas por la inflamación de los vasos sanguíneos localizados en la piel. Estos vasos pueden incluir capilares, vénulas, arteriolas, e incluso los vasos linfáticos. • La vasculitis cutánea puede manifestarse debido a diversas causas subyacentes. • Existe una amplia gama de presentaciones clínicas observables. • En una minoría de pacientes, esta condición se vincula con más, la artritis y la enfermedad renal. Los pacientes afectados pueden experimentar múltiples enfermedades autoinmunes simultáneamente.
¿Qué es la Vasculitis Cutánea? La vasculitis cutánea se define como un conjunto de afecciones caracterizadas por la inflamación de los vasos sanguíneos localizados en la piel. Estos vasos pueden incluir capilares, vénulas, arteriolas, e incluso los vasos linfáticos. • La vasculitis cutánea puede manifestarse debido a diversas causas subyacentes. • Existe una amplia gama de presentaciones clínicas observables. • En una minoría de pacientes, esta condición se vincula con más, la artritis y la enfermedad renal. Los pacientes afectados pueden experimentar múltiples enfermedades autoinmunes simultáneamente.
Las neoplasias malignas más frecuentemente observadas en asociación con el síndrome de Wiskott-Aldrich son la leucemia y el linfoma de células B.
Manifestaciones Cutáneas del Síndrome de Wiskott-Aldrich
En el síndrome de Wiskott-Aldrich se manifiestan diversas alteraciones cutáneas:
- Desarrollo de eccema agudo y crónico, clínicamente indistinguible del eccema atópico. La gravedad y la persistencia varían; en su forma más grave, este eccema es refractario a muchos tratamientos convencionales.
- Pueden aparecer petequias y hematomas, ya sea como consecuencia del rascado de la piel eccematosa o de manera espontánea.
- Las infecciones cutáneas oportunistas, como el molusco contagioso
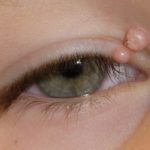 Comprendiendo el Molusco Contagioso: Causas, Contagio y Manifestaciones El molusco contagioso constituye una infección viral cutánea habitual, particularmente entre la población infantil, que se manifiesta a través de agrupaciones localizadas de pápulas epidérmicas, conocidas clínicamente como moluscos. ¿A quién afecta la infección por Molusco Contagioso? La infección por molusco contagioso afecta predominantemente a bebés y niños pequeños menores de 10 años. Su prevalencia es mayor en regiones con climas cálidos más, el herpes simple
Comprendiendo el Molusco Contagioso: Causas, Contagio y Manifestaciones El molusco contagioso constituye una infección viral cutánea habitual, particularmente entre la población infantil, que se manifiesta a través de agrupaciones localizadas de pápulas epidérmicas, conocidas clínicamente como moluscos. ¿A quién afecta la infección por Molusco Contagioso? La infección por molusco contagioso afecta predominantemente a bebés y niños pequeños menores de 10 años. Su prevalencia es mayor en regiones con climas cálidos más, el herpes simple Comprendiendo el Virus Herpes Simple: Definición y Transmisión El herpes simple constituye un trastorno infeccioso viral muy frecuente, manifestándose típicamente como una **erupción** o sensación abrasadora y localizada. La gran mayoría de la población experimentará una infección por este virus al menos una vez en su vida. Comúnmente, el herpes simple es reconocido por los nombres de herpes labial o **ampollas febriles**, debido a que los episodios recurrentes son a más y las bacterias, son comunes. La septicemia también puede manifestarse como un problema persistente o recurrente en pacientes con síndrome de Wiskott-Aldrich.
Comprendiendo el Virus Herpes Simple: Definición y Transmisión El herpes simple constituye un trastorno infeccioso viral muy frecuente, manifestándose típicamente como una **erupción** o sensación abrasadora y localizada. La gran mayoría de la población experimentará una infección por este virus al menos una vez en su vida. Comúnmente, el herpes simple es reconocido por los nombres de herpes labial o **ampollas febriles**, debido a que los episodios recurrentes son a más y las bacterias, son comunes. La septicemia también puede manifestarse como un problema persistente o recurrente en pacientes con síndrome de Wiskott-Aldrich.
Genética del Síndrome de Wiskott-Aldrich
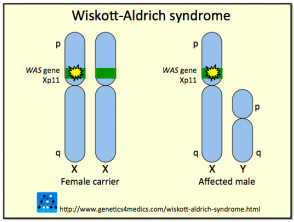
Síndrome de Wiskott Aldrich
* Imagen cortesía de Genetics 4 Medics
Pruebas Diagnósticas Clave para el Síndrome de Wiskott-Aldrich
En el hemograma completo, el recuento plaquetario es casi universalmente bajo, y las plaquetas exhiben característicamente un tamaño inferior al normal. Aunque en algunos casos el recuento plaquetario se encuentre dentro del rango normal, el tamaño plaquetario siempre se encuentra afectado. Asimismo, el recuento de neutrófilos y linfocitos puede resultar disminuido.
Si un lactante o niño con eccema presenta indicios de trombocitopenia y existe sospecha de inmunodeficiencia, debe realizarse un hemograma completo de inmediato. Si se confirma la trombocitopenia o se aprecian plaquetas diminutas en el frotis de sangre, se aconseja la derivación a un especialista pediátrico.
En casos de fuerte sospecha clínica del síndrome de Wiskott-Aldrich, y tras identificar anomalías en el hemograma, es fundamental consultar el caso con un inmunólogo pediátrico antes de proceder con investigaciones adicionales.
Inmunoglobulina (o anticuerpo) los niveles en el torrente circulatorio pueden ser reducidos en el síndrome de Wiskott-Aldrich. El síndrome clásico se relaciona con:
- Concentraciones bajas de inmunoglobulina M (IgM) e inmunoglobulina G (IgG).
- Niveles de inmunoglobulina A (IgA) e inmunoglobulina E (IgE) que oscilan entre normales y altos.
No obstante, especialmente en lactantes, estas alteraciones clásicas de las inmunoglobulinas podrían no ser evidentes, dado que el síndrome de Wiskott-Aldrich conlleva una disminución progresiva de la función inmunológica.
Las respuestas de anticuerpos a la vacunación pueden estar
el diagnóstico definitivo para el síndrome de Wiskott-Aldrich se establece mediante un análisis genético de sangre y linfocitos, el cual busca identificar mutaciones o deleciones específicas dentro del gen WASP, especialmente crucial cuando hay respuestas deficientes a las vacunas neumocócicas.
Opciones de Tratamiento para el Síndrome de Wiskott-Aldrich
El trasplante de célula madre hematopoyética, frecuentemente un trasplante de médula ósea, se considera el tratamiento curativo principal para los pacientes con síndrome de Wiskott-Aldrich. Para aquellos que tienen un hermano donante genéticamente compatible (un grupo que representa cerca del 20% de los pacientes), la tasa de supervivencia alcanza el 80%. La supervivencia es levemente menor, pero comparable, al utilizar un donante no emparentado compatible. Cuando se requiere un donante emparentado incompatible, la tasa de supervivencia disminuye y la incidencia de complicaciones aumenta. Es fundamental señalar que los resultados son significativamente mejores si el trasplante de células madre hematopoyéticas se realiza durante las primeras etapas de la infancia.
En las situaciones en las que el trasplante no es una opción o mientras se espera el procedimiento, los niños afectados requieren manejo específico para abordar la inmunodeficiencia y los problemas de sangrado. Estos tratamientos sintomáticos incluyen:
- Administración diaria de profilaxis con Trimetoprim-sulfametoxazol profilaxis para prevenir la infección por *Pneumocystis jiroveci*.
- Profilaxis con aciclovirAciclovir: Mecanismo de Acción y Pautas de Tratamiento Antiviral El aciclovir es reconocido como el antivírico más recetado a nivel mundial. Su disponibilidad inicial con prescripción médica se remonta a 1983. Químicamente, el aciclovir es un compuesto sintético cuya estructura molecular imita a los nucleósidos de purina. Su eficacia principal radica en su capacidad para inhibir la replicación del virus. Se ha comprobado que detiene el crecimiento del virus del más para la prevención de infecciones por el virus del herpes simple.
- Infusiones periódicas de inmunoglobulina intravenosaFundamentos de la Inmunoglobulina Intravenosa (IGIV): Definición y Composición La inmunoglobulina intravenosa (IGIV) se define como un producto biológico obtenido mediante la combinación del plasma de un gran grupo de donantes, típicamente entre 10,000 y 20,000 individuos. Este producto recibe un alto grado de purificación a través de un proceso de fraccionamiento utilizando alcohol frío. La composición predominante de la IGIV está constituida por el subtipo IgG de las inmunoglobulinas, más en casos de anomalías en la función de la Célula B.
- Transfusiones de plaquetas irradiadas y glóbulos rojos para controlar episodios hemorrágicos severos.
Es crucial evitar las vacunas vivas, como BCG y MMR, ya que están contraindicado. Asimismo, deben omitirse los medicamentos antiinflamatorio no esteroideos (AINEs), incluyendo ibuprofeno, diclofenaco y aspirina.
Actualmente, la terapia genética para tratar el síndrome de Wiskott-Aldrich se mantiene en fase experimental.
Pronóstico para Niños con Síndrome de Wiskott-Aldrich
Sin un trasplante de células madre hematopoyéticas, la esperanza de vida media para los niños diagnosticados con síndrome de Wiskott-Aldrich oscila entre los 15 y 20 años. Se espera que aquellos que reciben un injerto celular logren una supervivencia mucho más prolongada y mejoren su calidad de vida.
Información Genética Esencial para las Familias
Una vez que se identifica la anomalía genética en el niño afectado, es posible realizar pruebas a la madre para determinar si es portadora del gen mutado. Si se confirma que es portadora, existe un riesgo del 50% de tener futuros hijos afectados. Además, hay un 50% de probabilidad de transmitir el gen a sus hijas, quienes se convertirían en portadoras y podrían, a su vez, tener hijos afectados. Para las familias con historial conocido, el diagnóstico prenatal está disponible.


