Cos'è Prader - Willi sindrome?
La sindrome di Prader-Willi è la più comune genetico causa dell'obesità. Fu descritto per la prima volta nel 1887 da John Langdon Down, 70 anni prima di Prader et al nel 1956. È anche nota come sindrome di Prader-Labhart-Willi.
Sindrome di Prader-Willi

Eugenia Martínez Vallejo, vestita da Don Juan Carreño de Miranda (c.1680). Si ritiene che Eugenia h
* Credito: Museo Nazionale del Prado.
Chi ha la sindrome di Prader-Willi?
La sindrome di Prader-Willi si verifica circa una volta ogni 25.000 nati vivi, ma è probabilmente più comune perché la condizione non viene diagnosticata precocemente.
La sindrome di Prader-Willi ha autosomica eredità dominante (ereditata da un genitore affetto) e colpisce entrambi i sessi e tutte le razze. Tuttavia, la maggior parte dei casi è sporadica.
Qual è la causa della sindrome di Prader-Willi?
La sindrome di Prader-Willi deriva dalla mancanza di espressione del PWC regione di cromosoma 15. Il geni per la sindrome di Prader-Willi sono normalmente espresse solo sul cromosoma ereditato dal padre, e la copia del cromosoma 15 ereditata dalla madre è disabilitata.
Sono coinvolti tre meccanismi.
- In 75% dei casi, i geni Prader-Willi mancanti sono ereditati da un genitore non affetto (cioè, delezione paterna 15q11-q13).
- Nel 20%, entrambe le copie del cromosoma 15 sono ereditate dalla madre (cioè, disomia uniparentale materna [mUPD])
- In 5%, l'inattivazione di questi geni è dovuta a un difetto di imprinting nella regione critica di Prader-Willi del genitore del cromosoma 15.
La sindrome di Angelman è un disturbo clinicamente distinto che deriva da un difetto di imprinting di origine madre mappato sullo stesso cromosoma della sindrome di Prader-Willi.
Meccanismi genetici della sindrome di Prader-Willi *
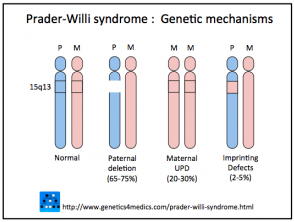
PraderWilli Genetics
* Immagine per gentile concessione di Genetics 4 Medics.
Quali sono le caratteristiche cliniche della sindrome di Prader-Willi?
Le caratteristiche cliniche della sindrome di Prader-Willi dipendono dall'età dell'individuo.
Infanzia
- Difficoltà ad alimentarsi e scarso riflesso di suzione
- Pianto diminuito o assente
- Sonnolenza
- Flaccidità
- Tardi presto evolutivo pietre miliari
- Mancato successo
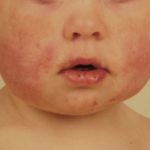
- Caratteristiche del viso tra cui occhi a mandorla, bocca rivolta verso il basso, una distanza ridotta tra le tempie e un labbro superiore sottile
- Ricoveri ospedalieri per torace infezioni.
Infanzia
- Comportamento di ricerca di cibo che porta all'obesità
- Bassa statura
- Capricci
- Un'alta soglia del dolore
- Disordini del sonno
- Ti prude la pelle
- Strabismo
- L'aspetto caratteristico del viso diventa più evidente
- Mani e piedi piccoli, con dita sottili e affilate
- Disabilità intellettuale
- Ricoveri ospedalieri per chirurgia.
L'età adulta
- Pubertà ritardata
- Testicoli piccoli o ritenuti con pieghe scrotali ridotte o rughe
- Ciclo mestruale assente / irregolare
- Ortopedico problemi, inclusi scoliosi o cifosi della colonna vertebrale; anca displasia; arti disallineati e gambe arcuate.
Complicanze ortopediche della sindrome di Prader-Willi
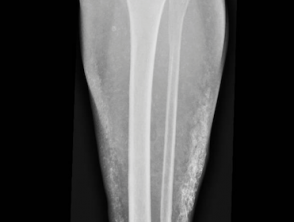
Osteoma cutaneo a placca acquisito
Quali sono i file cutaneo caratteristiche della sindrome di Prader-Willi?
Pelle
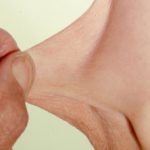
Pizzicare la pelle è molto comune ed è la caratteristica della pelle più tipica della sindrome di Prader-Willi. Lesioni sono presenti in tutte le fasi dell'evoluzione della sindrome. I segni includono graffi, sanguinamento, batterica pelle infezione, croste, cicatrici e secondarie milia - soprattutto sul dorso delle mani e degli avambracci.
Altre caratteristiche della pelle possono includere:
- Macchie marcate sulla pelle dei neonati
- Pelle di colore chiaro, capelli e occhi (tipo di pelle Fitzpatrick I-II)
- Smagliature addominali (smagliature) legate all'obesità / eccesso di cibo durante l'infanzia
- Erisipela, soprattutto nella fascia di età più avanzata
- Pannicolite
- Gonfiore alle gambe e edema con vene varicose
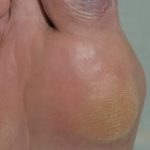
- Piezogenico papule, riportato in quattro pazienti su cinque esaminati
- Lividi a causa di un'elevata soglia del dolore
- Ipogonadismo, con conseguente riduzione o assenza dei peli del viso, delle ascelle e del pube, soprattutto negli uomini
- Spirali di capelli su precedente cuoio capelluto
- Segni cutanei di diabete mellito, incluso acantosi nigricans.
Sono disponibili anche segnalazioni di casi singoli di orticaria pelle pigmentata, estremamente secca, seborroico dermatitee pseudo-Kaposi sarcoma.
Bocca
Le caratteristiche della sindrome di Prader-Willi possono includere:
- Di spessore saliva sui bordi della bocca
- Denti piccoli con smalto sottosviluppato che causa la carie
- Palato alto e arcuato.
Anogenitale regione
Le caratteristiche della sindrome di Prader-Willi possono includere:
- Rettale pizzicando la pelle, con conseguente sanguinamento, ulcere, anemiae costipazione
- Vulva sottosviluppata labbra (labbra) nelle donne
- Riduzione delle rughe e delle pieghe scrotali nei maschi.
Condizioni della pelle che possono colpire le persone con sindrome di Prader-Willi
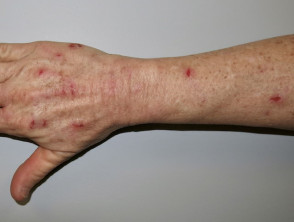
Ti prude la pelle
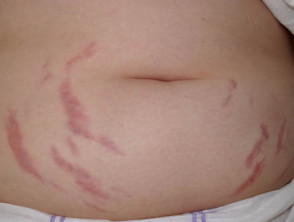
Smagliature addominali
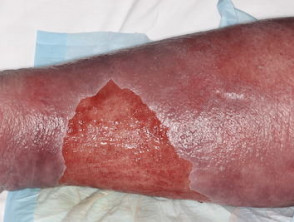
Erisipela
Come viene diagnosticata la sindrome di Prader-Willi?
I seguenti criteri diagnostici possono essere utilizzati per diagnosticare la sindrome di Prader-Willi.
Criteri principali
Ciascuno dei seguenti criteri ottiene 1 punto:
- Caratteristiche facciali caratteristiche: occhi a mandorla, bocca rivolta verso il basso, una distanza ridotta tra le tempie e un labbro superiore sottile
- Pietre miliari dello sviluppo ritardato
- Problemi alimentari o ritardo della crescita durante l'infanzia.
- Ipogonadismo (ipoattivo sesso ormoni)
- Floppy alla nascita
- Rapido aumento di peso tra 1 e 6 anni.
Criteri minori
Ciascuno dei seguenti criteri ottiene 1 punto:
- Diminuzione dei movimenti fetali e letargia infantile.
- Problemi agli occhi e alla vista, inclusi strabismo e miopia
- Pelle, capelli e colore degli occhi pallidi (rispetto ad altri membri della famiglia)
- Mani strette con bordo ulnare dritto
- Bassa statura (rispetto ad altri membri della famiglia)
- Ti prude la pelle
- Disturbi del sonno o Apnea notturna.
Ricerca
Gli studi genetici sono il modo più accurato per diagnosticare la sindrome di Prader-Willi. Questi studi devono essere considerati in ogni neonato molle che necessita di alimentazione mediante sonda. I tre meccanismi della sindrome devono essere ricercati in sequenza, a partire dalla delezione paterna.
I raggi X dovrebbero essere presi durante l'infanzia per identificare i problemi scheletrici, poiché questi possono essere mascherati dall'obesità.
Quali sono le complicazioni della sindrome di Prader-Willi?
Nella vita adulta, i problemi dermatologici che richiedono un trattamento sono un problema di salute significativo, con l'erisipela che è un motivo comune per il ricovero ospedaliero.
Le complicanze mediche della sindrome di Prader-Willi includono:
- Obesità patologica
- Ipertensione
- carriera
- Vena profonda trombosi (coaguli nelle gambe)
- Polmonare stantuffo (coaguli nei polmoni)
- Infarti
- Ansia
- Infezioni toraciche e polmonite.
- Diabete mellito di tipo 2
- Osteoporosi e osteopenia
- Modelli di consumo anormali che portano a intossicazione da acqua
- Incapacità di vomitare
Diagnosi differenziale Sindrome di Prader-Willi
La diagnosi differenziale della sindrome di Prader-Willi include altre cause di obesità e ritardo della crescita nell'infanzia e nell'infanzia.
Sindrome da delezione 6q16
Una sindrome simile a Prader-Willi da considerare è la sindrome da delezione 6q16, che è dovuta a prossimale delezioni del braccio lungo del cromosoma 6. Le sue caratteristiche principali sono:
- Obesità e alimentazione eccessiva.
- Ipotonia
- Mani e piedi piccoli
- Problemi agli occhi e alla vista
- Ritardo dello sviluppo globale.
Cosa comporta il trattamento della sindrome di Prader-Willi?
La consulenza genetica è consigliata per future gravidanze e familiari. Potrebbe essere necessaria un'attenzione particolare anestesia per le persone con sindrome di Prader-Willi. I trattamenti affrontano le complicazioni e le associazioni mediche e chirurgiche quando si presentano e sono correlate all'età dell'individuo colpito.
Infanzia
- Il trattamento può essere necessario per difficoltà di alimentazione e cedimento (p. Es., Alimentazione mediante sondino)
- Anche le infezioni respiratorie sono comuni in questa fascia di età.
Infanzia
- Le cause più comuni di ricovero ospedaliero nella sindrome di Prader-Willi sono correlate alla malattia (p. Es., Correzione ortopedica).
- L'amministrazione di inquietante Mangiare, che porta ad un aumento di peso eccessivo e ad altri disturbi comportamentali, diventa un grave problema per le famiglie.
Adolescenza
Possono essere richiesti ricoveri ospedalieri per:
- Problemi correlati agli ormoni (p. Es., Carenza di ormone della crescita)
- Chirurgia della scoliosi.
Adulti
Il ricovero ospedaliero può essere richiesto per:
- Chirurgia per a inguinale ernia
- Eventi medici correlati a diabete mellito e infezioni della pelle (p. Es., Erisipela)
- Psichiatrico Episodi
- Intossicazione da acqua o droghe.
I farmaci usati dagli adulti con sindrome di Prader-Willi includono psicotropi, lassativi, prodotti per la pelle e trattamenti per il diabete.
Anziani
Le infezioni respiratorie sono un grave problema di salute negli anziani.