
«`html
Aspectos Clave del Síndrome de Prader-Willi: Causas, Prevalencia y Manifestaciones
Definición y Contexto del Síndrome de Prader-Willi
El síndrome de Prader-Willi (SPW) se identifica como la causa genética más frecuente de obesidad. Curiosamente, John Langdon Down lo describió inicialmente en 1887, setenta años antes de que Prader et al. lo detallaran en 1956. Este trastorno también es conocido históricamente como síndrome de Prader-Labhart-Willi.
Representación Histórica del Síndrome de Prader-Willi

Eugenia Martínez Vallejo, vestida por Don Juan Carreño de Miranda (c. 1680). Se cree que Eugenia h
* Crédito: Museo Nacional del Prado.
Prevalencia: ¿Quiénes son afectados por el Síndrome de Prader-Willi?
Se estima que el síndrome de Prader-Willi afecta aproximadamente a 1 de cada 25,000 nacidos vivos. No obstante, la incidencia real podría ser superior, dada la subestimación frecuente por diagnósticos tardíos o no realizados.
El SPW se clasifica como una condición de herencia autosómico dominante (en términos de mecanismo de herencia afectado, aunque genéticamente es complejo por la impronta) y afecta a todas las etnias y géneros por igual. A pesar de este patrón heredable, la gran mayoría de los casos reportados son esporádicos.
Causas Genéticas Fundamentales del Síndrome de Prader-Willi
El síndrome de Prader-Willi surge de la ausencia de expresión génica en la región cromosoma 15 específica del PWC. Normalmente, los genes asociados al SPW solo se expresan desde la copia heredada del padre; la copia del cromosoma 15 recibida de la madre permanece silenciada (inactiva).
Existen tres mecanismos genéticos principales que conducen a esta alteración:
- En el 75% de los casos, la falta de expresión génica se produce porque la herencia de los genes de Prader-Willi proviene de un padre no afectado (esto se conoce como deleción paterna en la región 15q11-q13).
- Alrededor del 20% de los casos se deben a la herencia de ambas copias del cromosoma 15 de la madre (disomía uniparental materna [mUPD]).
- En el 5% restante, la inactivación génica es resultado de un defecto en el proceso de impronta en la región crítica del cromosoma 15 paterno.
Es fundamental notar que el síndrome de Angelman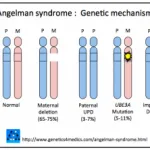 ¿Qué es el Síndrome de Angelman? El síndrome de Angelman es un trastorno neurológico poco frecuente que afecta aproximadamente a 1 de cada 15,000 nacimientos. Históricamente, se confundía con otras condiciones como la parálisis cerebral o el autismo, debido a su complejo formación de síntomas distintivos. Este síndrome fue nombrado en honor al Dr. Harry Angelman, quien describió la afección por primera vez en 1965. ¿A quién afecta el Síndrome más, un trastorno distinto, surge de defectos de impronta derivados de la madre mapeados en la misma región cromosómica que el SPW.
¿Qué es el Síndrome de Angelman? El síndrome de Angelman es un trastorno neurológico poco frecuente que afecta aproximadamente a 1 de cada 15,000 nacimientos. Históricamente, se confundía con otras condiciones como la parálisis cerebral o el autismo, debido a su complejo formación de síntomas distintivos. Este síndrome fue nombrado en honor al Dr. Harry Angelman, quien describió la afección por primera vez en 1965. ¿A quién afecta el Síndrome más, un trastorno distinto, surge de defectos de impronta derivados de la madre mapeados en la misma región cromosómica que el SPW.
Mecanismos Genéticos Implicados en el SPW *
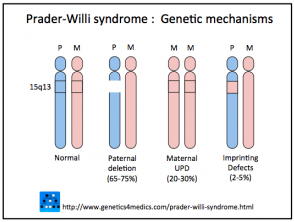
Genética de PraderWilli
* Imagen cortesía de Genetics 4 Medics.
Características Clínicas Distintivas del Síndrome de Prader-Willi
Las características clínicas del síndrome de Prader-Willi son complejas y varían con la edad, comenzando con hipotonía severa neonatal y dificultades de alimentación. A medida que los afectados crecen, las manifestaciones cambian radicalmente hacia una hiperfagia incontrolable, lo que impulsa la obesidad mórbida si no se gestiona. Además, suelen presentar características faciales específicas, déficit cognitivo leve a moderado, y problemas endocrinos.
```
Manifestaciones Clínicas del Síndrome de Prader-Willi Según la Edad
Las manifestaciones clínicas del síndrome de Prader-Willi varían significativamente dependiendo de la etapa de desarrollo del individuo, impactando distintas áreas de salud desde la infancia hasta la edad adulta.
Síntomas en la Infancia Temprana
Durante la etapa infantil, los signos iniciales del síndrome de Prader-Willi son frecuentemente asociados a dificultades de alimentación y tono muscular bajo:
- Dificultad marcada para alimentarse y un reflejo de succión deficiente.
- Llanto disminuido o incluso ausente.
- Somnolencia notable.
- Flacidez generalizada (hipotonía).
- Retraso en la adquisición de los hitos tempranos del desarrollo.
- Fracaso para aumentar de peso de manera adecuada.
- Rasgos faciales distintivos, como ojos amendrados, boca con comisuras caídas, distancia intercantal estrecha y labio superior delgado.
- Necesidad de ingresos hospitalarios debido a infecciones torácicas recurrentes.
Manifestaciones Durante la Infancia Posterior
A medida que el niño crece, la hiperfagia comienza a ser un síntoma dominante, llevando a problemas de peso y cambios conductuales:
- Comportamiento insistente de búsqueda de alimentos, lo cual conduce a la obesidad mórbida.
- Estatura baja en comparación con pares de edad similar.
- Episodios frecuentes de berrinches o rabietas.
- Umbral de dolor elevado, lo que puede dificultar la detección de lesiones.
- Trastornos del sueño, como apnea obstructiva del sueño.
- Hábito de pellizcarse la piel (skin picking).
- Estrabismo (desviación de la alineación de los ojos).
- La apariencia facial característica se vuelve más evidente.
- Manos y pies pequeños, acompañados de dedos delgados y puntiagudos.
- Discapacidad intelectual leve a moderada.
- Necesidad de hospitalizaciones para procedimientos quirúrgicos relacionados con otras complicaciones.
Características Clínicas en la Edad Adulta
En la adultez, las características se centran en las deficiencias hormonales y las complicaciones musculoesqueléticas:
- Pubertad retrasada o incompleta.
- Hipogonadismo: testículos pequeños o no descendidos en hombres, con pliegues o arrugas escrotales reducidas.
- Ausencia o irregularidad del ciclo menstrual en mujeres.
- Problemas ortopédicos, incluyendo la presencia de escoliosis o cifosis vertebral; displasia de cadera; mala alineación de las extremidades y desarrollo de piernas arqueadas.
Complicaciones Ortopédicas Asociadas al Síndrome de Prader-Willi
Osteoma cutis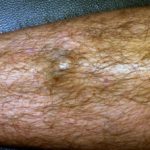 Osteoma Cutis: Definición, Incidencia y Características Clínicas ¿Qué es el Osteoma Cutis? El osteoma cutis se define como la formación anómala de hueso dentro de la estructura cutánea. Este tejido óseo generalmente se origina a partir de un foco de calcificación que surge de un proceso inflamatorio, una cicatriz o un granuloma subyacente. Es crucial destacar que el osteoma cutis es una condición benigna, es decir, no es canceroso. Esta más en placa adquirido.
Osteoma Cutis: Definición, Incidencia y Características Clínicas ¿Qué es el Osteoma Cutis? El osteoma cutis se define como la formación anómala de hueso dentro de la estructura cutánea. Este tejido óseo generalmente se origina a partir de un foco de calcificación que surge de un proceso inflamatorio, una cicatriz o un granuloma subyacente. Es crucial destacar que el osteoma cutis es una condición benigna, es decir, no es canceroso. Esta más en placa adquirido.
Características Cutáneas del Síndrome de Prader-Willi
Piel
El acto de pellizcarse la piel (skin picking) es una manifestación extremadamente frecuente y se considera la característica cutánea más representativa del síndrome de Prader-Willi. Estas lesiones se observan en todas las fases de la enfermedad e incluyen marcas de rascado, sangrado, infección bacteriana secundaria de la piel, formación de costras, cicatrices y aparición de milia, focalizándose especialmente en el dorso de las manos y los antebrazos.
Otras manifestaciones dermatológicas que pueden presentar los afectados incluyen:
- Marcado moteado de la piel presente desde el período neonatal.
- Coloración clara de la piel, el cabello y los ojos (generalmente tipo de piel Fitzpatrick I-II).
- Estrías
 ¿Qué son las estrías y sus tipos? Las estrías son fisuras o líneas finas que aparecen en la piel, resultado del desgarro del tejido subcutáneo provocado por el estiramiento rápido o excesivo de la misma. Aunque representan una condición muy común y generalmente no suponen un riesgo médico significativo, pueden generar preocupación estética en ciertas personas. Existen diversas denominaciones para referirse a las estrías, basadas en su apariencia, causa o más abdominales (estrías) vinculadas a la obesidad resultante del apetito insaciable durante la infancia.
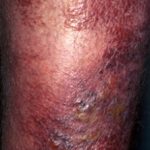
¿Qué son las estrías y sus tipos? Las estrías son fisuras o líneas finas que aparecen en la piel, resultado del desgarro del tejido subcutáneo provocado por el estiramiento rápido o excesivo de la misma. Aunque representan una condición muy común y generalmente no suponen un riesgo médico significativo, pueden generar preocupación estética en ciertas personas. Existen diversas denominaciones para referirse a las estrías, basadas en su apariencia, causa o más abdominales (estrías) vinculadas a la obesidad resultante del apetito insaciable durante la infancia. - ErisipelaComprendiendo la Erisipela: Causas, Síntomas y Factores de Riesgo Definición y Naturaleza de la Erisipela La erisipela se define como una variante superficial de la celulitis, caracterizada por ser una infección bacteriana potencialmente grave que afecta las capas superiores de la piel. Específicamente, esta afección compromete la dermis superior y se propaga a través de los cutáneos linfáticos superficiales. Históricamente, es conocida popularmente como el "fuego de San Antonio" debido más, más común en el grupo etario de mayor edad.
- PaniculitisEntendiendo la Paniculitis: Definición y Clasificación Detallada La paniculitis se define como un grupo de afecciones caracterizadas por la inflamación del tejido graso subcutáneo. Aunque sus etiologías son variadas, la manifestación clínica suele ser similar en la mayoría de los casos. El diagnóstico definitivo se establece mediante una biopsia cutánea, ya que la causa subyacente se revela a través de características microscópico específicas. Clínicamente, la forma más frecuente y conocida más.
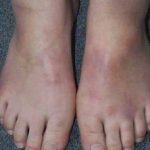
- Hinchazón de las piernas (edema) acompañada de várices.
- Pápulas piezogénicas, reportadas en aproximadamente el ochenta por ciento de los pacientes examinados.
- Hematomas fáciles debido al umbral de dolor elevado.
- Hipogonadismo, que se manifiesta como una reducción o ausencia de vello facial, axilar y púbico, siendo más notorio en varones.
- Patrón de cabello en espiral en la zona frontal del cuero cabelludo.
- Signos cutáneos asociados a la diabetes mellitus, como la acantosis nigricans.

Adicionalmente, se han documentado casos únicos de urticaria pigmentosa, piel extremadamente seca, dermatitis Comprendiendo la Dermatitis: Definición, Causas y Tipos Comunes La dermatitis abarca un conjunto de afecciones inflamatorias que se manifiestan a través de cambios específicos en la epidermis, manifestándose frecuentemente como picazón intensa. Esta condición es notablemente común, afectando a cerca de una quinta parte de la población en algún momento de sus vidas. Debido a su etiología diversa, la dermatitis presenta múltiples patrones de manifestación clínica. Los términos "dermatitis" y más seborreica y pseudo-Kaposi sarcoma.
Comprendiendo la Dermatitis: Definición, Causas y Tipos Comunes La dermatitis abarca un conjunto de afecciones inflamatorias que se manifiestan a través de cambios específicos en la epidermis, manifestándose frecuentemente como picazón intensa. Esta condición es notablemente común, afectando a cerca de una quinta parte de la población en algún momento de sus vidas. Debido a su etiología diversa, la dermatitis presenta múltiples patrones de manifestación clínica. Los términos "dermatitis" y más seborreica y pseudo-Kaposi sarcoma.
Boca
Las variaciones orales asociadas al síndrome de Prader-Willi pueden comprender:
- Producción de saliva espesa en los ángulos de la boca.
- Dientes de tamaño reducido con esmalte subdesarrollado, lo que aumenta la susceptibilidad a las caries dentales.
- Paladar alto y arqueado.
Región Anogenital
Las características encontradas en la región anogenital del síndrome de Prader-Willi incluyen:
- (El contenido de la lista original parecía truncado, se mantiene la estructura para completar la sección.)
- Pellizcarse repetitivamente la piel en el área rectal, lo que puede provocar sangrado, desarrollo de úlceras, anemia y estreñimiento crónico.
- Desarrollo insuficiente de la vulva (labios) en el caso de las mujeres.
- Observación de la reducción de pliegues y arrugas escrotales en los varones.
Afecciones dermatológicas asociadas al síndrome de Prader-Willi
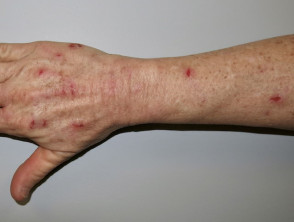
Pellizcado de piel
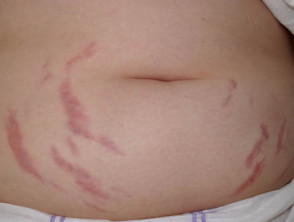
Estrías abdominales
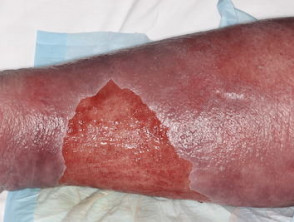
Erisipela
Procedimiento de diagnóstico para el síndrome de Prader-Willi
Para confirmar la presencia del síndrome de Prader-Willi, se emplean los siguientes criterios diagnósticos, los cuales se dividen en categorías principales y menores.
Criterios diagnósticos fundamentales
Cada uno de los siguientes indicadores aporta 1 punto para el diagnóstico:
- Presentación de rasgos faciales distintivos: ojos con forma almendrada, comisuras bucales orientadas hacia abajo, distancia reducida entre las sienes craneales, y un labio superior notablemente delgado.
- Retrasos significativos en la adquisición de hitos clave del desarrollo.
- Dificultades en la alimentación o un crecimiento lento durante la etapa infantil temprana.
- Diagnóstico de hipogonadismo (niveles bajos de hormonas sexuales).
- Flacidez muscular generalizada ("floppy bebé") al nacer.
- Rápido e excesivo incremento de peso corporal entre los 1 y los 6 años de edad.
Criterios diagnósticos secundarios
La presencia de cada uno de los siguientes aspectos suma 1 punto adicional:
- Disminución de los movimientos fetales durante la gestación y episodios de letargo durante la infancia.
- Problemas oculares y visuales existentes, incluyendo la tendencia a entrecerrar los ojos o la aparición de miopía.
- Coloración pálida de la piel, el cabello y los ojos en comparación con otros familiares directos.
- Observación de manos estrechas que presentan un borde cubital de aspecto recto.
- Estatura final por debajo del promedio esperable para la edad y el sexo familiar.
- Hábito persistente de pellizcarse la piel de forma autodirigida.
- Alteraciones en los patrones de sueño o diagnóstico de apnea del sueño.
Investigaciones y confirmación
La metodología más fiable para diagnosticar el síndrome de Prader-Willi reside en la realización de estudios genéticos detallados. Estos análisis son imperativos, especialmente en recién nacidos hipotónicos que requieren soporte nutricional mediante sonda. La búsqueda de los tres mecanismos genéticos causales debe realizarse de forma secuencial, iniciando siempre por la deleción paterna.
Asimismo, es fundamental solicitar radiografías durante los años de crecimiento para poder detectar cualquier anomalía esquelética, dado que estas condiciones pueden quedar ocultas o ser difuminadas por el desarrollo de la obesidad.
¿Cuáles son las complicaciones asociadas al Prader-Willi?
Complicaciones y Manejo del Síndrome de Prader-Willi
En la etapa adulta, las afecciones dermatológicas que demandan intervención médica representan una preocupación significativa para la salud, siendo la erisipela un motivo habitual de hospitalización.
Complicaciones Médicas Asociadas al Síndrome de Prader-Willi
Las complicaciones médicas que pueden surgir debido al síndrome de Prader-Willi son diversas e impactan múltiples sistemas corporales:
- Obesidad mórbida
- Hipertensión arterial
- Accidentes cerebrovasculares (ACV)
- Vena profunda trombosis (formación de coágulos en las piernas)
- Pulmonar émbolo (coágulos que viajan a los pulmones)
- Eventos cardíacos isquémicos (ataques al corazón)
- Trastornos de ansiedad
- Infecciones respiratorias graves y neumonía.
- Diabetes mellitus tipo 2
- Osteoporosis y osteopenia (baja densidad ósea)
- Patrones de ingesta hídrica inadecuados que pueden conducir a intoxicación por agua
- Incapacidad fisiológica para inducir el vómito.
Diagnóstico Diferencial del Síndrome de Prader-Willi
Al realizar el diagnóstico diferencial del síndrome de Prader-Willi, es fundamental considerar otras etiologías que presenten obesidad y retraso del crecimiento durante la infancia y niñez.
Síndrome de Deleción 6q16
El síndrome de deleción 6q16 se presenta como un cuadro clínico similar al de Prader-Willi, resultado de deleciones proximales en el brazo largo del cromosoma 6. Sus manifestaciones principales incluyen:
- Obesidad y patrón de hiperfagia o alimentación excesiva.
- Hipotonía muscular generalizada.
- Extremidades (manos y pies) de tamaño reducido.
- Problemas oculares y visuales.
- Retraso significativo en el desarrollo global.
Estrategias de Tratamiento para el Síndrome de Prader-Willi según la Etapa Vital
Se aconseja la asesoría genética, especialmente para futuros embarazos y para informar a los demás miembros de la familia. Además, las personas diagnosticadas con síndrome de Prader-Willi pueden requerir precauciones especiales en relación con la anestesia. El tratamiento se centra en gestionar las complicaciones médicas y quirúrgicas a medida que estas se manifiestan, adaptándose a la edad específica del individuo afectado.
Etapa Infantil
- Puede ser necesaria intervención para manejar las dificultades de alimentación y la flacidez (p. ej., soporte mediante alimentación por sonda).
- Las infecciones broncopulmonares son frecuentes en esta cohorte etaria.
Etapa Escolar (Infancia Tardía)
- Las causas más comunes de internamiento hospitalario en este grupo están asociadas a patologías ortopédicas o sistémicas (p. ej., procedimientos de corrección ortopédica).
- El manejo del apetito insaciable, conocido como comer obsesionante, que conlleva un aumento de peso descontrolado y otras alteraciones conductuales, se convierte en un desafío primario para las familias.
Adolescencia
Durante la adolescencia, pueden ser necesarios ingresos hospitalarios para atender:
- Cuestiones hormonales y endocrinológicas (p. ej., diagnóstico y manejo de la deficiencia de la hormona del crecimiento).
- Cirugía correctiva para la escoliosis.
Adultez
En la vida adulta, la hospitalización puede ser necesaria por los siguientes motivos:
- Procedimientos quirúrgicos para corregir una inguinal hernia.
- Manejo de eventos médicos secundarios a diabetes mellitus o infecciones cutáneas (notablemente la erisipela).
- Descompensaciones o episodios psiquiátricos.
- Tratamiento de emergencias por intoxicación relacionada con ingesta excesiva de agua o sustancias.
Los adultos con síndrome de Prader-Willi a menudo requieren medicaciones que abarcan psicotrópicos, laxantes, productos tópicos para el cuidado de la piel y tratamientos específicos para controlar la diabetes.
Personas Mayores
En los adultos mayores, las infecciones respiratorias recurrentes o graves constituyen una morbilidad significativa que requiere seguimiento activo.
El abordaje integral del síndrome de Prader-Willi exige una vigilancia multidisciplinaria constante, ajustando los cuidados médicos según la progresión y las necesidades específicas de cada fase del desarrollo de la persona afectada.


