Che cos'è Ehlers - Danlos? sindrome?
La sindrome di Ehlers-Danlos (EDS) è un gruppo di malattie ereditarie che coinvolgono a genetico difetto collagene o tessuto connettivo sintesi e struttura [1]. Questo risulta in:
- Pelle fragile e iperelastica
- Articolazioni instabili e iperestensibili (ipermobile)
- Tessuto fragile e vasi sanguigni.
Sindrome di Ehlers-Danlos
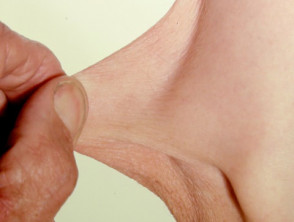
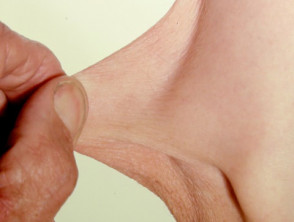
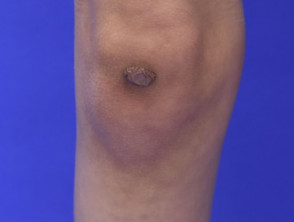
Iperelasticità della pelle del gomito
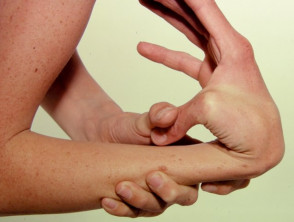
Ipermobilità
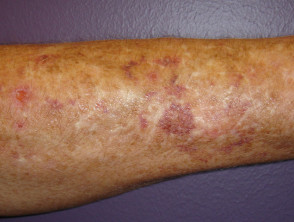
Lividi e cicatrici
Chi ha la sindrome di Ehlers-Danlos?
La frequenza complessiva dell'EDS è di circa 1 su 5000. L'EDS può verificarsi negli uomini e nelle donne di tutte le razze e di solito si manifesta per la prima volta nell'infanzia e nella vita dei giovani adulti.
EDS ha autosomica dominante e autosomica recessiva modelli di ereditarietà.
- il autosomica dominante La malattia si verifica quando solo una copia di un file gene è necessario per causare una condizione. C'è una probabilità 50% che una prole erediti il gene anomalo da un genitore affetto.
- La malattia autosomica recessiva richiede due copie di un gene anormale per causare la malattia; quindi un bambino di due anni vettore I genitori sono a rischio per 25% e 50% di sviluppando malattia ed essere portatore di una malattia, rispettivamente.
Sottotipi della sindrome di Ehlers-Danlos
Secondo la classificazione internazionale 2017 per le sindromi di Ehlers-Danlos, esistono tredici sottotipi di EDS, classificati in base alle caratteristiche cliniche, al modello di ereditarietà e molecolare difetti genetici.
Ognuno di loro è un disturbo diverso che "è vero" in una famiglia. Ciò significa che i membri di una famiglia affetta da EDS condivideranno le stesse caratteristiche. Alcuni casi non si adattano perfettamente a un tipo noto di EDS e un paziente può mostrare caratteristiche di più di un tipo. Clinico dimostrazioni e la sua gravità può variare in modo significativo, anche all'interno della stessa famiglia.
Hypermobile EDS è il sottotipo più comune di EDS (MIM 130020), seguito da Classic EDS (MIM 130000) e vascolare EDS (MIM 130050). Gli altri tipi di EDS sono molto rari.
Vedi sottotipi di sindrome di Ehlers-Danlos [visualizza la tabella in una nuova scheda] [2,3]
Quali sono le cause della sindrome di Ehlers-Danlos?
Il collagene è uno dei principali elementi costitutivi del corpo. È un proteina Si trova ampiamente nella struttura di molti tessuti e organi del corpo. Esistono diversi tipi di collagene, ciascuno con proprietà diverse. Il collagene può fornire forza e sostegno deciso, può essere elastico per consentire il movimento o può essere usato per tenere insieme le cose.
Un difetto genetico può causare quantità ridotte di collagene, interruzione del collagene (il collagene è solitamente disposto in fasci) e alterazioni nelle dimensioni e nella forma del collagene. Il sottotipo di EDS dipende da come il collagene metabolismo è stato colpito. Ad esempio, l'EDS vascolare è causato dalla sintesi del collagene di tipo III ridotta o assente.
Ci sono alcuni tipi di EDS che sono il risultato di altri la matrice extracellulare disturbi e loro componenti (come i glicosaminoglicani) e difetti in intracellulare In lavorazione.
Quali sono le caratteristiche cliniche della sindrome di Ehlers-Danlos?
Segni e sintomi differiscono per tipo e gravità tra i diversi tipi di EDS. Di seguito è riportato un riepilogo delle caratteristiche cliniche dell'EDS basato sui sistemi.
Caratteristiche facciali
- Un naso sottile e stretto
- Occhi prominenti
- Orecchie senza lobi
Coinvolgimento parodontale
- Grave e intrattabile infiammazione gomma
- Mancanza di gomma appiccicosa
Muscoloscheletrico sistema
- Ipermobilità: le articolazioni si piegano più del solito. Le dita sono le più colpite, ma tutte le articolazioni possono essere colpite. Congenita o ricorrente Le lussazioni o sublussazioni articolari possono verificarsi soprattutto nelle spalle, nelle anche e nelle ginocchia.
- Talipes congenito equinovaro (piede torto) - a evolutivo deformità del piede dove uno o entrambi i piedi sono rivolti verso l'interno e verso il basso.
- Cifoscoliosi congenita o ad esordio precoce: una curvatura anormale della colonna vertebrale.
- Aracnodattilia: dita delle mani e dei piedi anormalmente lunghe e sottili
- Congenita o lieve, insorgenza tardiva di ipotonia e ritardo motore lordo
Ipermobilità nella sindrome di Ehlers-Danlos
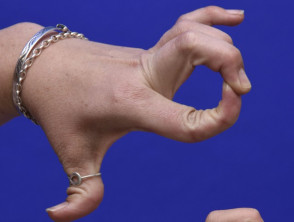
Articolazioni ipermobile
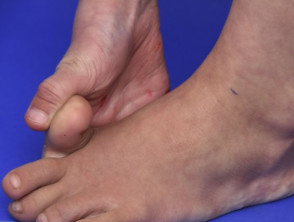
Articolazioni ipermobile
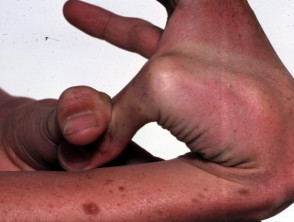
Articolazioni ipermobile
Pelle
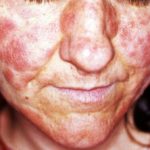
- Iperestensibilità della pelle: è facile separare la pelle dal corpo e, una volta rilasciata, si ritrae al suo stato originale.
- Slim e traslucido pelle con vasi sanguigni sottostanti visibili.
- Pelle fragilità - la pelle si rompe e si strappa facilmente.
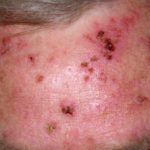
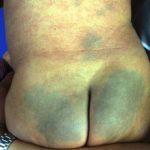
- Lividi e lividi può insorgere dopo lesioni banali e può essere visto in siti non soggetti a trauma.
- Aspetto prematuramente invecchiato di pelle sottile e rugosa, soprattutto su mani e piedi.
- Atrofico cutaneo Cicatrici - Le ferite guariscono molto lentamente, provocando cicatrici nella bocca del pesce o nella carta di sigaretta.
- Pieghe epicantiche far sembrare largo il ponte del naso.
- Pseudotumori dei molluschi: si tratta di piccole escrescenze spugnose di 2 o 3 cm di diametro su punti di pressione come ginocchia e gomiti.
- Sottocutaneo sferoidi: lobi di grasso calcificati che sono il risultato di un insufficiente apporto di sangue.
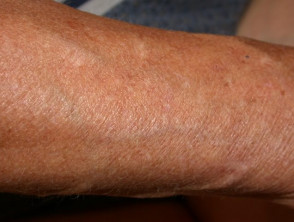
- Noduli piccoli grumi sodi appena sotto la superficie della pelle che appaiono più spesso sulle braccia e sugli stinchi.
- Pretibiale piatti.
Iperestensibilità nella sindrome di Ehlers-Danlos
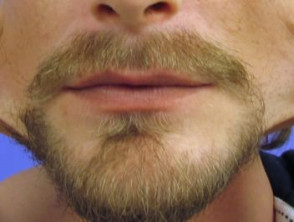
Iperelasticità della pelle delle guance.
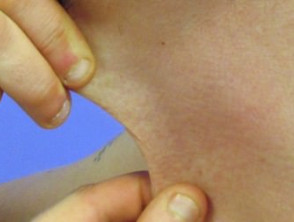
Iperelasticità della pelle del collo.
Iperelasticità della pelle del gomito
Lividi nella sindrome di Ehlers-Danlos
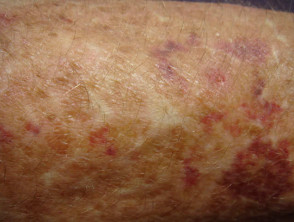
Sindrome di Ehlers-Danlos
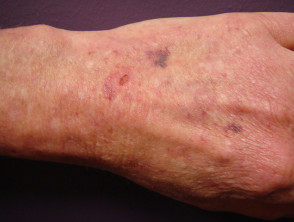
Sindrome di Ehlers-Danlos
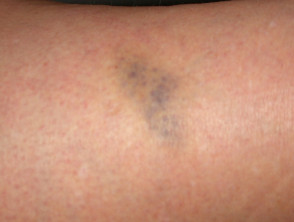
Lividi
Cicatrici atrofiche nella sindrome di Ehlers-Danlos
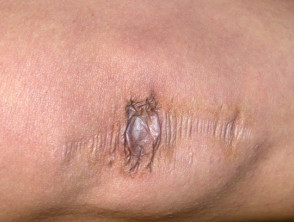
Cicatrici estese in un paziente con sindrome di Ehlers-Danlos.
Cicatrice atrofica
Cicatrici ipopigmentate
Sindrome cutanea di Ehlers-Danlos
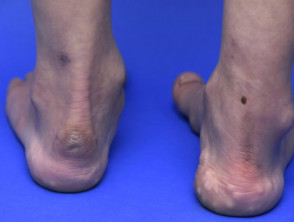
Pseudotumore alla caviglia
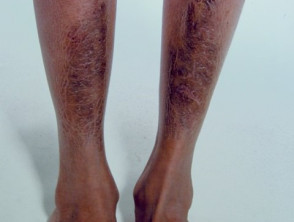
Placche pretibiali
Guarda altre immagini della sindrome di Ehlers-Danlos.
Occhio
- magro cornea, con o senza interruzione
- blu sclerotico
- Cheratocono: la cornea è a forma di cono
- Keratoglobus - il globulare rigonfiamento e assottigliamento della cornea
Difetti interni del collagene
- Soffio cardiaco (prolasso della valvola mitrale) e pareti intestinali indebolite. arteriee utero, che può essere rotto
- Autonomo disfunzione - transitorio perdita di coscienza, palpitazioni, vertigini, dolore toracico e mancanza di respiro
- Ortostatica posturale tachicardia sindrome (POTS): battito cardiaco accelerato, che si verifica in particolare quando si cambia postura o in piedi
- Ricorrente pneumotorace - questa potrebbe essere la presentazione iniziale di EDS.
Quali sono le complicazioni della sindrome di Ehlers-Danlos?
Possibili complicazioni della sindrome di Ehlers-Danlos includono:
- Cronico dolori articolari
- Scarsa guarigione delle ferite
- Fallimento della chiusura della ferita chirurgica
- Inizio precoce arterite
- Rottura prematura della membrana fetale
- Rottura di organi vascolari e cavi
- Rottura del palloncino, distacco della retina e glaucoma
- Epilessia [4].
Come viene diagnosticata la sindrome di Ehlers-Danlos?
Il sospetto di sindrome di Ehlers-Danlos dovrebbe portare a un'anamnesi e un esame accurati. Criteri principali e minori sono stati sviluppati per ciascuno dei 13 sottotipi di EDS [3]. La diagnosi definitiva di una variante specifica richiede tessuto biopsia e l'analisi genetica molecolare dovuta alla varietà di difetti molecolari e alla sovrapposizione clinica tra i sottotipi di EDS.
Storia medica
Storia personale e familiare dettagliata dei muscoli, oculare, pelle, malattie dentali, cardiache e respiratorie.
Esame clinico
- Esame completo della pelle - Valuta l'iperestensibilità della pelle e le cicatrici anormali.
- Il Beighton scala - valutare l'ipermobilità di un'articolazione, in cui un punteggio di 5 o più indica molto diffuso ipermobilità articolare.
Ricerca
- Biopsia cutanea per istologia, fibroblasti culturaed elettrone microscopia
- Quando si sospetta EDS cifoscoliotica, viene prelevato un campione di urina per misurare il rapporto tra i legami crociati della lisil-piridinolina e l'idrossilisil-piridinolina.
I test genetici molecolari confermano la diagnosi, tranne nel caso di EDS ipermobile, che ha una base genetica sconosciuta e quindi è una diagnosi clinica che deve soddisfare tutti i criteri clinici.
Qual è diagnosi differenziale per la sindrome di Ehlers-Danlos?
Le condizioni che possono essere scambiate per EDS includono:
- Osteogenesi imperfetta
- Sindrome di Loeys-Dietz
- Displasie scheletriche
- Mucopolisaccaridosi
- Pelle sindrome di laxa
- Pseudoxantoma elastico
- Ulrich muscolare congenito distrofia
- Bethlem miopatia
- Sindrome di Larsen
- Acondroplasia
- Sindrome RIN2
- Sindrome di Marfan
- Condrodisplasia
- Miopatie congenite
- Sindrome di Freeman-Sheldon.
Qual è il trattamento per la sindrome di Ehlers-Danlos?
Le linee guida di gestione dipendono dal sottotipo EDS.
- Adotta misure preventive contro complicazioni gravi e pericolose per la vita.
- I pazienti dovrebbero considerare di indossare un braccialetto MedicAlert per comunicare il loro stato EDS.
- Tutti i pazienti con EDS dovrebbero essere indirizzati per consulenza genetica e specialisti appropriati.
Misure generali
- I bambini possono trarre beneficio da assorbenti e bende protettive.
- Contatta sport o attività che coinvolgono intracranica La pressione (come suonare la tromba) dovrebbe essere evitata per evitare traumi indebiti.
- Il paziente può trarre vantaggio da esercizi a bassa resistenza e terapia fisica.
- Considera gli integratori di calcio e vitamina D per ottimizzare la densità ossea.
- Fornire supporto psicologico dopo la diagnosi.
- I pazienti devono indossare un braccialetto medico di avvertenza con la scritta "tipo EDS".
Medicinale
- L'acido ascorbico (vitamina C) è stato utilizzato per ridurre i lividi.
Chirurgia e cura delle ferite
- La chirurgia deve essere eseguita con cautela dato il rischio di complicanze vascolari.
- Le ferite devono essere chiuse accuratamente con suture prive di tensione.
- Si consigliano punti profondi; lasciare suture superficiali per periodi prolungati per consentire la guarigione.
La gravidanza
- Il monitoraggio ostetrico è consigliato durante tutta la gravidanza a causa del rischio di sanguinamento e disturbi vascolari, chirurgici e anestetico complicazioni.
- Le donne con EDS corrono un rischio maggiore di parto pretermine e postpartum emorragia, prolasso vescicale e uterino, ernie addominali e deiscenza della ferita.
- Non ci sono linee guida per la gestione ostetrica dell'EDS data l'ampia gamma di possibili implicazioni e la gravità di ogni sottotipo; pertanto, i pazienti vengono trattati caso per caso.
Screening e sorveglianza
- Il paziente deve avere un monitoraggio regolare della pressione sanguigna e arterioso screening delle malattie per ridurre il rischio di sanguinamento e complicanze vascolari.
- No-invasivo mettere sullo schermo - ecografia, tomografia computerizzata (Connecticut) scansione, ecocardiogrammae risonanza magnetica (Risonanza magnetica) può essere fatto per identificare possibili complicazioni.
- Le procedure di screening invasivo devono essere considerate attentamente.
- Le scansioni della densità ossea a doppia energia per assorbimetria a raggi X (DEXA) devono essere eseguite ogni pochi anni.
- I pazienti con KEDS dovrebbero sottoporsi regolarmente a un esame della vista poiché sono a rischio di complicazioni oculari.
Qual è il risultato per i pazienti con sindrome di Ehlers-Danlos?
il previsione dei pazienti con sindrome di Ehlers-Danlos varia notevolmente anche all'interno dello stesso sottotipo. La maggior parte dei pazienti ha un'aspettativa di vita e una funzione cognitiva normali. Le persone con EDS vascolare possono avere un'aspettativa di vita più breve e un aumentato rischio di morte improvvisa a causa della rottura di un organo interno e di un vaso principale.