Cos'è Muckle-Wells? sindrome?
La sindrome di Muckle-Wells (MWS, MIM 191900) è rara genetico autoinfiammatorio sindrome e la forma di gravità intermedia della sindrome periodica associata alla criopirina (CAPS). Come con altre forme di questa sindrome, si verifica con ricorrente episodi di febbre, pelle eruzione, dolori articolari, dolori addominali e congiuntivite, ma in aggiunta, i pazienti di solito sviluppare un progressivo neurosensoriale sordità e amiloidosi.
Famiglia La sindrome autoinfiammatoria da freddo (FCAS) e NOMID / CINCA sono condizioni strettamente correlate. Famiglie e individui sono stati segnalati con MWS innescata dall'esposizione al freddo (una caratteristica di FCAS) e che mostrano anche lievi caratteristiche NOMID / CINCA, come mal di testa mattutino, bassa statura, prominenza della fronte, appiattimento del ponte nasale o papilledema (gonfiore del disco ottico nell'occhio).
Chi ha la sindrome di Muckle-Wells e perché?
La sindrome di Muckle-Wells colpisce prevalentemente gli europei e si presenta prima dei 20 anni.
La sindrome di Muckle-Wells viene ereditata come autosomica condizione dominante, il che significa che ogni figlio di una vittima ha una probabilità di 50% sviluppando sindrome. I casi senza una storia familiare sono raramente identificati. Questo può metà un padre è un asintomatico vettore del mutazione, si è verificata una mutazione spontanea o non è stata identificata alcuna mutazione in DNA sequenziamento.
Molecolare biologia e genetica
il proteina colpita nella sindrome di Muckle-Wells è la criopirina, prodotta dal NLRP3 gene Situato in cromosoma 1. Il gene è espresso nei globuli bianchi (principalmente neutrofili) e condrociti (cellule della cartilagine). La criopirina è una componente essenziale di inflammasome, a intracellulare complesso proteico coinvolto in innato sistema immunitario. L'inflammasoma anormale nella sindrome di Muckle-Wells consente l'attivazione illimitata del enzima caspase-1, che a sua volta causa una sovrapproduzione di asset interleuchina-1β, accendendo il infiammatorio cascata in modo incontrollato.
La stessa mutazione puntiforme può essere identificata nella MWS e nella sindrome autoinfiammatoria del freddo familiare (FCAS): R260W, V198M e V262G; e meno frequentemente in MWS con lieve NOMID / CINCA.
Quali sono le caratteristiche cliniche della sindrome di Muckle-Wells?
il febbrile Gli episodi della sindrome di Muckle-Wells sono molto simili a quelli osservati nella sindrome familiare associata al freddo (FCAS), ma persistono da 1 a 3 giorni (o più a lungo) e senza la stretta e definita associazione con l'esposizione al freddo. Tipicamente, la sindrome di Muckle-Wells è associata sviluppo di ipoacusia neurosensoriale progressiva.
Le caratteristiche cliniche di acuto Gli attacchi della sindrome di Muckle-Wells possono includere:
- Febbre: non presente in tutti i casi, ma quando presente è solitamente più alta che in FCAS (39-40 ° C)
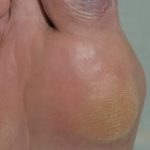
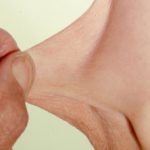
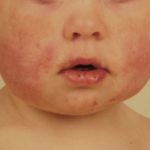
- Acne - orticaria-come Ma atipico
- Dolore articolare: varia da brevi episodi di artralgia un gonfiore ricorrente di grandi articolazioni con possibile distruzione articolare o poliarticolare artrite. Può essere aggravato dall'esercizio fisico e trauma. Dopo che le ossa smettono di crescere, è stato segnalato un miglioramento dell'artralgia. Questo può essere associato a dolore muscolare (mialgia).
- Congiuntivite - comune
- Altre condizioni infiammatorie agli occhi, episclerite, uveite e iridociclite.
- Mal di testa: grave, indica asettico meningite o sollevato intracranica Pressione.
- La fatica può essere persistente e grave, influendo sulla qualità della vita
- Dolore addominale - segnalato durante le riacutizzazioni
- Bocca ulcere.
Le caratteristiche sovrapposte con FCAS o NOMID / CINCA sono spesso presenti nella sindrome di Muckle-Wells quando ricercate. Il freddo come fattore scatenante per gli attacchi, frequenti mal di testa e papilledema asintomatico (gonfiore nella parte posteriore dell'occhio) sono alcuni esempi. Anche all'interno di una famiglia con la stessa mutazione genetica, le caratteristiche possono variare da MWS "puro" in un membro a sovrapporsi in un altro e FCAS "puro" in un terzo.
Cosa fa scattare gli attacchi?
I sintomi possono peggiorare verso la notte. Alcuni pazienti hanno episodi acuti rari intermittenti mentre altri hanno sintomi quotidiani o continui. La durata degli attacchi è generalmente più lunga rispetto a FCAS e dura da diversi giorni a continui.
La maggior parte dei pazienti non identifica alcun trigger. Stress fisico ed emotivo, stanchezza e infezione è informato a precipitato attacchi acuti. Gli episodi febbrili possono essere innescati dal freddo, sovrapponendosi a FCAS.
Progresso
Progressivo bilaterale La sordità neurosensoriale si sviluppa in due terzi dei pazienti con sindrome di Muckle-Wells e di solito compare per la prima volta dall'infanzia alla prima età adulta. La perdita ad alta frequenza è la prima cartello. È stato suggerito questo cronica infiammazione nella coclea dell'orecchio interno provoca la perdita permanente di Corti capelli cellule (e perdita dell'organo di Corti) e cocleari nervo danneggiare.
L'amiloidosi complica la sindrome di Muckle-Wells non trattata in 251 pazienti TP1T, che inizia nella vita adulta come proteinuria, progredisce fino a insufficienza renale e morte entro 10 anni. Periferica neuropatia A causa dell'amiloidosi può svilupparsi più tardi dell'insufficienza renale.
La pubertà può essere ritardata, specialmente con malattie gravi.
Come viene diagnosticata la sindrome di Muckle-Wells?
La sindrome di Muckle-Wells dovrebbe essere considerata nelle caratteristiche cliniche tipiche, anche in assenza di una storia familiare positiva di sindrome periodica associata alla criopirina (CAPS).
Durante un attacco acuto, i test rivelano caratteristiche aspecifiche, tra cui:
- Reagenti di fase acuta (analisi del sangue): elevati eritrociti velocità di sedimentazione (ESR), proteina C-reattiva (CRP), siero amiloide A (SAA)
- Anemia malattia cronica
- Leucocitosi (numero elevato di globuli bianchi)
- Liquido cerebrospinale ad alta pressione (CSF) con neutrofili e eosinofili e senza infezione.
I seguenti test possono essere eseguiti in qualsiasi momento.
- Audiogramma: inizialmente perdita dell'udito ad alta frequenza, progressiva verso la sordità neurosensoriale bilaterale.
- Proteina urinaria per rilevare l'amiloidosi: il primo segno è una proteina nelle urine, senza globuli rossi o bianchi.
- Rene biopsia conferma la presenza di depositi di amiloide.
- I test genetici possono essere eseguiti utilizzando un test commerciale a NLRP3 mutazione genetica nell'esone 3.
Qual è il trattamento per la sindrome di Muckle-Wells?
Anakinra, un'interleuchina-1 ricevitore antagonista, è un agente biologico che è stato utilizzato per la prima volta nel 2003 per trattare due pazienti non correlati alla sindrome di Muckle-Wells con un effetto miracoloso sugli episodi acuti. Febbre, eruzione cutanea e congiuntivite hanno risposto entro 4 ore e i marker ematici dell'infiammazione sono tornati alla normalità entro una settimana. Successivamente è stato utilizzato per trattare con successo molti pazienti con sindrome di Muckle-Wells.
Sebbene ci siano state segnalazioni di un miglioramento della sordità, altre hanno mostrato un peggioramento continuo. Forse, l'inversione della perdita dell'udito può essere correlata al tempo trascorso dall'esordio, essendo utile un intervento più precoce rispetto al trattamento tardivo. Nei bambini piccoli trattati prima dello sviluppo della perdita dell'udito, sembra che questa complicanza sia prevenibile. È necessario un follow-up più lungo per determinarlo.
Anakinra ha anche dimostrato di invertire l'amiloide dichiarazione in alcuni casi.
Molti casi gravi hanno avuto sintomi lievi residui di MWS con follow-up a lungo termine. Questi potrebbero aver risposto a dosi più elevate. I bambini piccoli hanno bisogno di una dose giornaliera più alta per kg di peso corporeo rispetto ai bambini più grandi e agli adulti controllo sintomi. L'aumento del volume di iniezione può essere associato a un aumento del disagio locale e del dolore nel sito di iniezione. L'iperattività è stata segnalata con il successo del trattamento, forse a causa del miglioramento della fatica. Può verificarsi un aumento di peso, in particolare nelle donne. È richiesto un trattamento quotidiano indefinito, con rapido ricomparsa sintomi nei giorni successivi cessazione di iniezioni.
Nota: anakinra non è registrato o sovvenzionato in Nuova Zelanda (marzo 2011). In altri paesi come gli Stati Uniti e l'Europa, la sua indicazione registrata è l'artrite reumatoide.
Due antagonisti dell'interleuchina-1 sono approvati dalla FDA per il trattamento della sindrome di Muckle-Wells. Rilonacept è stato approvato per il trattamento della MWS negli adulti e nei bambini di età pari o superiore a 12 anni. Rilonacept è una proteina di fusione che impedisce all'interleuchina-1 di legarsi al suo recettore. Viene somministrato settimanalmente come sottocutaneo iniezione.
Canakinumab è stato approvato dalla FDA per il trattamento di adulti e bambini di età pari o superiore a 4 anni affetti dalla sindrome di Muckle-Wells. È un'azione lunga monoclonale anticorpo specificamente diretto contro l'interleuchina-1β. Canakinumab viene somministrato mediante iniezione sottocutanea ogni 8 settimane. Lo sviluppo di Vertigine Durante questo trattamento è stato segnalato in particolare nei pazienti con MWS con sordità neurosensoriale.
La qualità della vita è notevolmente migliorata con il trattamento anti-interleuchina-1 per i pazienti con sindrome di Muckle-Wells. Il trattamento permanente deve essere iniziato il prima possibile per ridurre il rischio di sordità e amiloidosi.