¿Qué es Muckle-Wells? síndrome?
El síndrome de Muckle-Wells (MWS, MIM 191900) es una rara genético autoinflamatorio síndrome y la forma de gravedad intermedia del síndrome periódico asociado a criopirina (CAPS). Como ocurre con otras formas de este síndrome, se presenta con recurrente episodios de fiebre, piel erupción, dolor en las articulaciones, dolor abdominal y conjuntivitis, pero además, los pacientes suelen desarrollar una progresivo neurosensorial sordera y amilosis.
Familiar El síndrome autoinflamatorio por frío (FCAS) y NOMID / CINCA son afecciones estrechamente relacionadas. Se han informado familias e individuos con MWS desencadenado por la exposición al frío (una característica de FCAS) y que también muestran características leves de NOMID / CINCA, como dolores de cabeza matutinos, baja estatura, prominencia de la frente, aplanamiento del puente nasal o papiledema (hinchazón de el disco óptico en el ojo).
¿Quién contrae el síndrome de Muckle-Wells y por qué?
El síndrome de Muckle-Wells afecta predominantemente a europeos y se presenta antes de los 20 años.
El síndrome de Muckle-Wells se hereda como autosómico condición dominante, lo que significa que cada hijo de una víctima tiene un 50% de posibilidades de desarrollando el síndrome. Rara vez se identifican casos sin antecedentes familiares. Esto puede media un padre es un asintomático portador del mutación, se ha producido una mutación espontánea o no se ha identificado ninguna mutación en ADN secuenciación.
Molecular biología y genética
los proteína afectado en el síndrome de Muckle-Wells es la criopirina, producida a partir del NLRP3 gene localizado en cromosoma 1. El gen se expresa en glóbulos blancos (principalmente neutrófilos) y condrocitos (células de cartílago). La criopirina es un componente esencial del inflamasoma, un intracelular complejo proteico involucrado en el innato sistema inmunitario. El inflamasoma anormal en el síndrome de Muckle-Wells permite la activación ilimitada del enzima caspasa-1, que a su vez provoca una sobreproducción de activos interleucina-1β, encendiendo el inflamatorio cascada de manera incontrolada.
La misma mutación puntual se puede identificar en MWS y síndrome autoinflamatorio familiar por frío (FCAS): R260W, V198M y V262G; y con menos frecuencia en MWS con NOMID / CINCA leve.
¿Cuáles son las características clínicas del síndrome de Muckle-Wells?
los febril Los episodios del síndrome de Muckle-Wells son muy similares a los observados en el síndrome familiar asociado al frío (FCAS), pero persisten durante 1 a 3 días (o más) y sin la asociación estrecha y definida con la exposición al frío. Característicamente, el síndrome de Muckle-Wells se asocia con la desarrollo de la hipoacusia neurosensorial progresiva.
Las características clínicas de la agudo Los ataques del síndrome de Muckle-Wells pueden incluir:
- Fiebre: no está presente en todos los casos, pero cuando está presente suele ser más alta que en FCAS (39-40C)
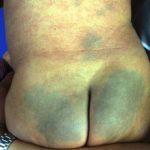
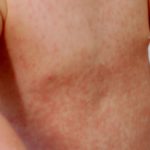
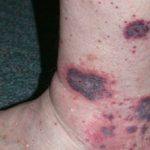
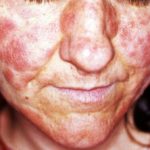
- Erupción cutanea - urticaria-como pero atípico
- Dolor articular: varía desde episodios cortos de artralgia a hinchazón recurrente de grandes articulaciones con posible destrucción articular o poliarticular artritis. Puede agravarse con el ejercicio y la trauma. Se ha informado una mejoría de la artralgia después de que los huesos dejan de crecer. Esto puede estar asociado con dolor muscular (mialgia).
- Conjuntivitis - frecuente
- Otras afecciones inflamatorias de los ojos, epiescleritis, uveítis e iridociclitis.
- Dolores de cabeza: severos, indican aséptico meningitis o levantado intracraneal presión.
- Fatiga - puede ser persistente y severa, impactando en la calidad de vida
- Dolor abdominal - informado durante los brotes
- Boca úlceras.
Las características de superposición con FCAS o NOMID / CINCA a menudo están presentes en el síndrome de Muckle-Wells cuando se buscan. El frío como desencadenante de ataques, dolores de cabeza frecuentes y papiledema asintomático (hinchazón en la parte posterior del ojo) son algunos ejemplos. Incluso dentro de una familia con la misma mutación genética, las características pueden variar desde MWS 'puro' en un miembro a superponerse en otro, y FCAS 'puro' en un tercero.
¿Qué desencadena los ataques?
Los síntomas pueden empeorar hacia la noche. Algunos pacientes tienen episodios agudos raros intermitentes mientras que otros tienen síntomas diarios o continuos. La duración de los ataques es generalmente más larga que para FCAS y dura desde varios días hasta continuos.
La mayoría de los pacientes no identifican ningún factor desencadenante. Estrés físico y emocional, fatiga y infección se informa a precipitado ataques agudos. Los episodios febriles pueden desencadenarse por frío, superponiéndose con FCAS.
Progreso
Progresivo bilateral La sordera neurosensorial se desarrolla en dos tercios de los pacientes con síndrome de Muckle-Wells, y por lo general aparece primero en la niñez hasta la edad adulta temprana. La pérdida de alta frecuencia es la primera firmar. Se ha sugerido que crónico inflamación en la cóclea del oído interno da como resultado la pérdida permanente de Corti cabello células (y pérdida del órgano de Corti) y coclear nervio dañar.
La amiloidosis complica el síndrome de Muckle-Wells no tratado en el 25% de los pacientes, comenzando en la vida adulta como proteinuria, progresando a insuficiencia renal y muerte en 10 años. Periférico neuropatía debido a la amiloidosis puede desarrollarse más tarde que la insuficiencia renal.
La pubertad puede retrasarse, especialmente con una enfermedad grave.
¿Cómo se diagnostica el síndrome de Muckle-Wells?
El síndrome de Muckle-Wells debe considerarse en las características clínicas típicas, incluso en ausencia de antecedentes familiares positivos de síndrome periódico asociado a criopirina (CAPS).
Durante un ataque agudo, las pruebas revelan características inespecíficas, que incluyen:
- Reactantes de fase aguda (análisis de sangre): elevados eritrocito velocidad de sedimentación (ESR), Proteína C-reactiva (CRP), suero amiloide A (SAA)
- Anemia de enfermedad crónica
- Leucocitosis (recuento alto de glóbulos blancos)
- Líquido cefalorraquídeo (LCR) a alta presión con neutrófilos y eosinófilos y sin infección.
Las siguientes pruebas se pueden realizar en cualquier momento.
- Audiograma: inicialmente hipoacusia de alta frecuencia, que progresa a sordera neurosensorial bilateral.
- Proteína urinaria para detectar la amiloidosis: el primer signo es una proteína en la orina, sin glóbulos rojos o blancos.
- Riñón biopsia confirma la presencia de depósitos de amiloide.
- Las pruebas genéticas se pueden realizar utilizando una prueba comercial para NLRP3 mutación genética en el exón 3.
¿Cuál es el tratamiento del síndrome de Muckle-Wells?
Anakinra, una interleucina-1 receptor antagonista, es un agente biológico que se utilizó por primera vez para tratar a dos pacientes no relacionados con el síndrome de Muckle-Wells en 2003 con un efecto milagroso sobre los episodios agudos. La fiebre, la erupción cutánea y la conjuntivitis respondieron en 4 horas y los marcadores sanguíneos de inflamación se normalizaron en una semana. Posteriormente se ha utilizado para tratar con éxito a muchos pacientes con síndrome de Muckle-Wells.
Aunque ha habido algunos informes de una mejora en la sordera, otros han mostrado un deterioro continuo. Posiblemente, la reversión de la pérdida auditiva puede estar relacionada con el tiempo transcurrido desde el inicio, siendo útil una intervención más temprana en comparación con el tratamiento tardío. En los niños pequeños tratados antes del desarrollo de la pérdida auditiva, parece que esta complicación se puede prevenir. Se requiere un seguimiento más prolongado para determinar esto.
También se ha demostrado que Anakinra revierte el amiloide declaración en algunos casos.
Muchos casos graves han tenido síntomas leves residuales de MWS con seguimiento a largo plazo. Estos pueden haber respondido a dosis más altas. Los niños pequeños necesitan una dosis diaria más alta por kg de peso corporal que los niños mayores y los adultos para controlar síntomas. El aumento del volumen de inyección puede estar asociado con un aumento de la incomodidad local y el dolor en el lugar de la inyección. Se ha informado hiperactividad con un tratamiento exitoso, quizás debido a la mejora de la fatiga. Puede ocurrir aumento de peso, particularmente en mujeres. Se requiere tratamiento diario indefinido, con rápido reaparición de síntomas en los días siguientes cesación de inyecciones.
Nota: anakinra no está registrada ni subvencionada en Nueva Zelanda (marzo de 2011). En otros países como EE.UU. y Europa, su indicación registrada es la artritis reumatoide.
Dos antagonistas de la interleucina-1 cuentan con la aprobación de la FDA para el tratamiento del síndrome de Muckle-Wells. Rilonacept ha sido aprobado para el tratamiento de MWS en adultos y niños a partir de los 12 años. Rilonacept es una proteína de fusión que evita la unión de la interleucina-1 a su receptor. Se administra semanalmente como subcutáneo inyección.
Canakinumab ha sido aprobado por la FDA para el tratamiento de adultos y niños a partir de los 4 años afectados por el síndrome de Muckle-Wells. Es una acción prolongada monoclonal anticuerpo dirigido específicamente contra la interleucina-1β. Canakinumab se administra mediante inyección subcutánea cada 8 semanas. El desarrollo de vértigo durante este tratamiento se ha informado particularmente en pacientes MWS con sordera neurosensorial.
La calidad de vida mejora notablemente con el tratamiento anti-interleucina-1 para pacientes con síndrome de Muckle-Wells. El tratamiento de por vida debe iniciarse lo antes posible para reducir el riesgo de sordera y amiloidosis.