Cos'è Legius sindrome?
La sindrome di Legius è rara genetico disturbo che è stato descritto per la prima volta nel 2007 [1]. Conosciuto anche come neurofibromatosi sindrome di tipo 1 [2].
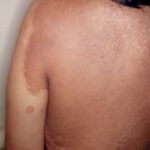
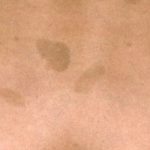
La sindrome di Legius è classicamente caratterizzata da multipli macule, noto come macule café au lait [3]. A differenza della neurofibromatosi di tipo 1, nella sindrome di Legius non si trovano tumori.
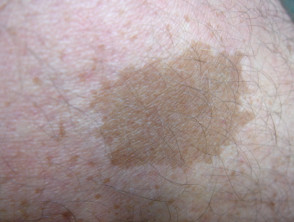
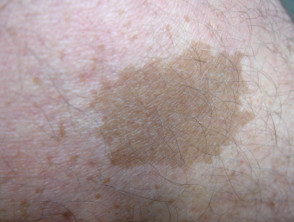
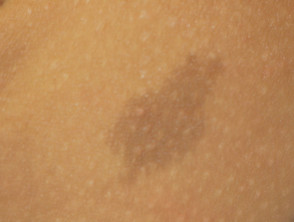
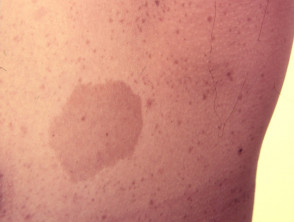
Caffè con macule di latte
Café au lait macule
Café au lait macule
Café au lait macule
Chi ha la sindrome di Legius?
La sindrome di Legius può verificarsi in chiunque abbia almeno un genitore biologico con sindrome di Legius geneticamente confermata [2]. il predominanza La sindrome di Legius è sconosciuta, ma può essere superiore al previsto a causa di una diagnosi errata della neurofibromatosi di tipo 1 [4].
Quali sono le cause della sindrome di Legius?
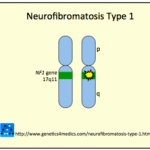
La sindrome di Legius è una condizione genetica ereditata in a autosomica modo dominante che coinvolge a PRIMAVERA1 gene mutazione in cromosoma 15q14. Questa mutazione provoca una funzione anormale di SPRED1 proteina, che è responsabile della regolazione delle specifiche vie di segnalazione cellulare coinvolte proliferazione, differenziazionee apoptosi. Nessun altro patogeno È stato scoperto che le varianti causano la sindrome di Legius. [2,3].
Come un autosomica dominante condizione, la sindrome di Legius può verificarsi in chiunque abbia almeno un genitore biologico con sindrome di Legius geneticamente confermata. In rari casi, può anche verificarsi de novo, ma questi casi non sono stati sufficientemente valutati per confermarlo [2]. Ogni bambino con un genitore con sindrome di Legius geneticamente confermata ha un rischio 50% di ereditare la condizione.
Quali sono le caratteristiche cliniche della sindrome di Legius?
Le caratteristiche cliniche della sindrome di Legius variano in modo significativo per natura e gravità da persona a persona.
Quasi tutti i pazienti con sindrome di Legius hanno più macule caffè-latte [2,3]. Il numero di queste macule tende ad aumentare durante l'infanzia. [4].
Altro comune cutaneo le caratteristiche possono includere:
- Lentiggini sotto le ascelle e inguinale regioni
- Lipomi [2–4].
Le caratteristiche non cutanee possono includere:
- Macrocefalia
- Caratteristiche facciali insolite, simili alla sindrome di Noonan
- Bassa statura
- Pectus excavatum (sterno infossato) o pectus carinatum (sterno sporgente) [2-4].
Le caratteristiche neuropsichiatriche possono includere:
- Difficoltà di apprendimento
- Sviluppo ritardare
- Disturbo da deficit di attenzione e iperattività (ADHD) [2-4].
Le disabilità intellettive sono generalmente meno gravi nei pazienti con sindrome di Legius rispetto ai pazienti con neurofibromatosi di tipo 1 [5].
È importante sottolineare che la sindrome di Legius non causa neurofibromi e Lisch noduli, che sono tipicamente osservati nella neurofibromatosi di tipo 1 [2,3].
Come viene diagnosticata la sindrome di Legius?
La sindrome di Legius è difficile da diagnosticare sulla base dei soli sintomi clinici, data la sua presentazione clinica cutanea simile ad altri disturbi con macule multiple di café-du-lait.
Quando si sospetta la sindrome di Legius, è possibile utilizzare test genetici per confermare la diagnosi. Questo implica:
- Analisi di sequenza PRIMAVERA1
- Analisi di cancellazione / duplicazione (se l'analisi della sequenza non è notevole)
- Un pannello di più geni per PRIMAVERA1 e un altro geni di interesse [2,3].
I risultati sospetti che possono giustificare i test genetici per la sindrome di Legius includono:
- Macule caffè au lait senza altre caratteristiche cliniche, suggerendo una neurofibromatosi di tipo 1
- Un padre con macule caffè au lait senza caratteristiche cliniche, che suggerisce ancora una volta una neurofibromatosi di tipo 1 [2].
Qual è diagnosi differenziale per la sindrome di Legius?
La sindrome di Legius viene spesso diagnosticata erroneamente perché è pigmentario dimostrazioni sono molto simili a quelli visti in altre sindromi con più lentigos. Una diagnosi corretta è essenziale per guidare il controllo e la gestione a lungo termine.
Neurofibromatosi di tipo 1
- La neurofibromatosi di tipo 1 è la diagnosi errata più comune a causa dei suoi criteri diagnostici clinici simili [2-4].
- Neurofibromi, nodi di Lisch, ossa anomaliee gliomi ottici non si verificano nella sindrome di Legius.
- Queste due condizioni sono difficili da differenziare nei bambini più piccoli, come i tumori caratteristici osservati nella neurofibromatosi generalmente non lo fanno. sviluppare fino a tarda età.
Sindrome di Noonan con più lentiggini
- La sindrome di Noonan con lentigine multipla (precedentemente nota come sindrome LEOPARD, coinvolge anomalie cardiovascolari, dell'orecchio e dei genitali, che non si verificano nella sindrome di Legius) [6].
Altre sindromi caratterizzate da più lentigo includono:
- Autosomica dominante macule di latte
- Sindrome di McCune-Albright.
Qual è il trattamento per la sindrome di Legius?
I bambini con sindrome di Legius dovrebbero sottoporsi a screening e valutazioni regolari per verificare ritardi nello sviluppo, declino cognitivo e problemi comportamentali. [2].
Il trattamento della sindrome di Legius è principalmente di supporto e dovrebbe concentrarsi sui problemi specifici dell'individuo colpito.
Una gestione appropriata, se indicata, può includere:
- Terapia fisica, del linguaggio e / o occupazionale
- Terapia di modificazione del comportamento e terapia farmacologica (cioè per l'ADHD) [2].
Qual è il risultato della sindrome di Legius?
I pazienti con sindrome di Legius hanno una buona previsione con una corretta gestione basata sui problemi.
Un individuo affetto deve considerare il rischio 50% di trasmissione genetica a ciascun bambino.