Síndrome de Majeed: Descripción Clínica y Fundamentos Genéticos
El síndrome de Majeed (MIM609628) se identifica como una enfermedad hereditaria sumamente infrecuente, catalogada dentro de las afecciones autoinflamatorias. Este trastorno se manifiesta durante los primeros años de vida a través de dos manifestaciones clínicas predominantes: la osteomielitis multifocal crónica y recurrente (CRMO, caracterizada por la inflamación ósea persistente), y la anemia diseritropoyética congénita (CDA), resultante de una producción deficiente de glóbulos rojos por parte de la médula ósea.
Adicionalmente, es frecuente observar brotes de inflamación cutánea de naturaleza neutrofílica, cuyos síntomas a menudo simulan el cuadro clínico de la enfermedad de Sweet. En estos episodios, el análisis mediante biopsia revela una alta acumulación de neutrófilos, definidos como glóbulos blancos polimorfonucleares.
Base Genética y Etiología del Síndrome de Majeed
El síndrome de Majeed representa una patología genética transmitida con un patrón de herencia autosómica recesiva. Esta modalidad de herencia implica que el individuo afectado debe haber heredado dos copias del gen mutado, recibiendo una copia defectuosa de cada progenitor, los cuales funcionan como portadores.
El responsable molecular de esta condición es el gen LPIN2, ubicado específicamente en la región del cromosoma 18p11.31, el cual codifica para la proteína Lipina 2. La investigación ha identificado tanto pacientes homocigotos —que poseen dos copias idénticas de la mutación— como heterocigotos compuestos —que presentan dos mutaciones diferentes afectando el mismo gen. Entre las alteraciones documentadas se encuentran mutaciones sin sentido y aquellas que causan errores en la secuencia (sentido erróneo).
Se calcula que la incidencia de estas alteraciones genéticas se sitúa aproximadamente en 1 de cada 35,000 nacimientos entre determinados grupos étnicos específicos, aunque no se ha confirmado su presencia en otros linajes. Resulta relevante señalar que mutaciones sin sentido en este mismo gen se han asociado previamente con casos de psoriasis. La Lipina 2 desempeña una función como enzima vital en el proceso de metabolismo de los lípidos (grasas). Si bien el camino patogénico preciso aún se investiga, la hipótesis principal sugiere una desregulación significativa del sistema inmunitario innato.
Hasta la fecha actual, el número de familias diagnosticadas con el síndrome de Majeed es reducido, concentrándose todos los casos confirmados en la región de Medio Oriente. Esta alta prevalencia de portadores entre las poblaciones árabes sustenta la postulación de que la condición está considerablemente subdiagnosticada en la práctica clínica habitual.
Las manifestaciones clínicas de esta enfermedad tienden a desarrollarse durante la infancia temprana, presentándose generalmente antes de cumplir los dos años de edad. El caso de inicio sintomático más precoz reportado se documentó a la asombrosa edad de 3 semanas de vida.
Manifestaciones Clínicas Detalladas del Síndrome de Majeed
Manifestaciones Clínicas y Diagnóstico del Síndrome de Majeed
Clínicamente, el síndrome de Majeed se caracteriza por brotes recurrentes de dolor articular e hinchazón, periódicamente acompañados de fiebre. Estos episodios suelen tener una duración de varios días y se presentan con una notable frecuencia, oscilando entre uno y tres ataques cada mes.
Las manifestaciones cardinales que distinguen el síndrome de Majeed incluyen la tríada sintomática siguiente:
- Osteomielitis multifocal crónica y recurrente.
- Anemia diseritropoyética congénita.
- Inflamación cutánea (transitoriamente neutrofílica).
Es crucial identificar estas características combinadas de manera temprana para establecer un diagnóstico preciso del síndrome de Majeed e iniciar el manejo terapéutico optimizado, el cual debe enfocarse en controlar la inflamación sistémica y abordar activamente la anemia subyacente.
El patrón clínico del síndrome presenta varios rasgos distintivos, especialmente en comparación con otras osteomielitis crónicas:
- Inicio de los síntomas en etapas tempranas de la vida.
- Reporte de dolor intenso en las áreas afectadas.
- Hinchazón y sensibilidad notables alrededor de las articulaciones comprometidas.
- Afectación principal de las articulaciones de mayor tamaño, si bien las articulaciones menores no están excluidas.
Diferenciación con CRMO y Pronóstico del Síndrome de Majeed
Distinguir el síndrome de Majeed de la Osteomielitis Multifocal Crónica Recurrente (CRMO) se basa en las siguientes diferencias observadas:
- La frecuencia de los episodios es significativamente mayor (al menos de forma mensual).
- Se establece como una condición que persiste a lo largo de toda la vida del paciente.
- La remisión espontánea es infrecuente y de muy corta duración.
- Provoca un retraso significativo e impactante en el crecimiento esperado.
- Las contracturas articulares se erigen como una complicación habitual y limitante.
Anemia Diseritropoyética Congénita Asociada
La anemia asociada al síndrome de Majeed muestra las siguientes características hematológicas:
- Se manifiesta como una anemia microcítica hipocrómica (hematíes pequeños y pálidos, con cantidad reducida).
- Su aparición se sitúa durante el transcurso del primer año de vida.
- La severidad muestra un amplio espectro, pudiendo variar desde un estado leve hasta ser completamente dependiente de transfusiones sanguíneas regulares.
Manifestaciones Dermatológicas
Las afecciones cutáneas no están presentes en todos los casos, pero cuando ocurren, son notables:
- La inflamación cutánea es transitoria y no constante.
- Puede presentarse como una dermatosis neutrofílica que imita la apariencia de la enfermedad de Sweet.
- Existe la posibilidad de desarrollar psoriasis (los portadores genéticos pueden manifestar esta condición).
- Presencia de Pustulosis palmoplantar.
- Manifestación de pustulosis cutánea.
- Acné
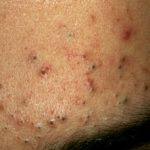 Dominando el Conocimiento: ¿Qué es el Acné y Cómo se Manifiesta? El acné es un trastorno crónico que afecta primariamente al folículo piloso y a la glándula sebácea. Esta condición se caracteriza por la dilatación y obstrucción de estos folículos, acompañada frecuentemente de inflamación. Existen diversas manifestaciones y variantes de esta afección cutánea. ¿Quiénes son Susceptibles de Padecer Acné? El acné tiene un alcance universal, afectando a hombres y mujeres más como posible síntoma asociado.
Dominando el Conocimiento: ¿Qué es el Acné y Cómo se Manifiesta? El acné es un trastorno crónico que afecta primariamente al folículo piloso y a la glándula sebácea. Esta condición se caracteriza por la dilatación y obstrucción de estos folículos, acompañada frecuentemente de inflamación. Existen diversas manifestaciones y variantes de esta afección cutánea. ¿Quiénes son Susceptibles de Padecer Acné? El acné tiene un alcance universal, afectando a hombres y mujeres más como posible síntoma asociado.
Características Clínicas Adicionales y Secuelas Óseas
Se han documentado otras características clínicas menos frecuentes en algunos pacientes:
- Desarrollo de hepatomegalia (aumento del tamaño del hígado).
- Presentación de Ictericia colestática neonatal, indicativa de una obstrucción temporal del conducto biliar en el periodo de recién nacido.
La cronicidad de la afectación ósea resulta en secuelas ortopédicas y de crecimiento observables a largo plazo:
- Retraso en la maduración de la edad ósea cronológica.
- Alcanzar una estatura adulta significativamente reducida.
- Desarrollo de contracturas de flexión que se vuelven permanentes.
La calidad de vida del individuo afectado puede verse considerablemente mermada debido a la naturaleza crónica de la enfermedad:
- Episodios de dolor recurrentes y debilitantes.
- Gestión constante de la anemia crónica.
- Limitaciones funcionales derivadas de las contracturas articulares.
- Desarrollo de Atrofia muscular secundaria a la inmovilización o falta de uso.
Es relevante mencionar que los individuos simplemente portadores del gen mutado (sin desarrollar la enfermedad completa) podrían manifestar únicamente las características cutáneas, como psoriasis o pustulosis; sin embargo, el número de estos casos observados es aún insuficiente para establecer una confirmación definitiva.
Criterios Diagnósticos y Confirmación Genética del Síndrome de Majeed
El diagnóstico diferencial del Síndrome de Majeed, dado su solapamiento sintomático con la Osteomielitis Multifocal Crónica Recurrente (CRMO), se establece rigurosamente mediante la concurrencia de criterios específicos:
- Demostrar la presencia de al menos 2 lesiones óseas características confirmadas radiográficamente.
- La sintomatología activa debe haberse mantenido durante un mínimo de 6 meses.
- Obtención de hallazgos histológicos consistentes mediante la biopsia de la lesión ósea.
- El diagnóstico etiológico debe establecerse antes de que el paciente alcance los 18 años de edad.
La confirmación definitiva del Síndrome de Majeed se logra al identificar mutaciones patogénicas en el gen LPIN2 a través de pruebas genéticas moleculares. Hasta el momento, esta representa la única alteración génica conocida y firmemente asociada a los casos reportados del Síndrome de Majeed.
Evaluación Diagnóstica Detallada del Síndrome de Majeed
El diagnóstico del Síndrome de Majeed se establece mediante la correlación de hallazgos clínicos con pruebas especializadas. A continuación, se detallan las conclusiones obtenidas de los estudios radiológicos y de laboratorio, fundamentales para diferenciarlo de otros procesos inflamatorios óseos.
Hallazgos en Pruebas de Imagen y Laboratorio
Evaluación Radiológica: Las radiografías evidencian lesiones típicas en las metáfisis de los huesos largos. Estas se manifiestan como áreas radiolúcidas de aspecto irregular (signo de osteólisis), típicamente circundadas por un margen de mayor densidad ósea, conocido como esclerosis.
Estudios de Medicina Nuclear: Al utilizar gammagramas óseos (con trazadores como Tc-99 o Ga-67), se observa una hipercaptación significativa en las lesiones que se encuentran activas o en proceso inflamatorio. Estos estudios son clave, ya que permiten detectar lesiones que aún permanecen asintomáticas en etapas tempranas.
Resonancia Magnética (RM): Esta modalidad diagnóstica se reputa como la más sensible para la identificación precisa de lesiones óseas activas, especialmente cuando estas afectan la estructura vertebral.
Biopsia Ósea: El análisis histológico revela un patrón de inflamación no infecciosa, caracterizado principalmente por la presencia de granulocitos, diferenciándolo de la osteomielitis bacteriana.
Biopsia de Médula Ósea: En el tejido medular, se observa un aumento en la producción de glóbulos rojos (eritropoyesis), incluyendo signos de diseritropoyesis, manifestada por la presencia de normoblastos binucleados y trinucleados.
Biopsia de Piel ```html Guía Completa sobre la Biopsia de Piel: Definición y Tipos ¿Qué es una Biopsia de Piel y Cómo se Realiza? Una biopsia de piel implica la extracción minuciosa de una muestra de tejido cutáneo para su posterior análisis. Generalmente, este procedimiento se realiza bajo anestesia local. Se administra una inyección del anestésico directamente en el área a tratar, lo cual puede causar una sensación punzante transitoria. Finalizado el muestreo, más: Cuando la manifestación cutánea está presente, el estudio histológico identifica abscesos neutrofílicos localizados intraepidérmicos.
```html Guía Completa sobre la Biopsia de Piel: Definición y Tipos ¿Qué es una Biopsia de Piel y Cómo se Realiza? Una biopsia de piel implica la extracción minuciosa de una muestra de tejido cutáneo para su posterior análisis. Generalmente, este procedimiento se realiza bajo anestesia local. Se administra una inyección del anestésico directamente en el área a tratar, lo cual puede causar una sensación punzante transitoria. Finalizado el muestreo, más: Cuando la manifestación cutánea está presente, el estudio histológico identifica abscesos neutrofílicos localizados intraepidérmicos.
Cultivos: Es importante destacar que los cultivos tomados de diversas fuentes (hueso, médula ósea, sangre y piel) resultan uniformemente negativos, lo cual respalda el diagnóstico diferencial excluyendo etiologías infecciosas.
Análisis de Sangre: Los resultados hematológicos suelen incluir:
- Una anemia que se clasifica como hipocrómica y microcítica.
- Persistencia de niveles elevados en la velocidad de sedimentación de los eritrocitos (VSG), siendo este un hallazgo constante.
- El conteo total de glóbulos blancos podría permanecer dentro del rango normal o presentar leucocitosis leve.
Resulta fundamental distinguir este síndrome del cuadro conocido como Osteomielitis Multifocal Recurrente Crónica (CRMO, MIM 259680). Aunque el CRMO puede tener componentes hereditarios, su inicio es generalmente más tardío en la infancia (entre los 4 y 14 años) y su presentación clínica más habitual es esporádica.
Los episodios sintomáticos en el Síndrome de Majeed tienden a presentarse con menor frecuencia, mostrando periodos de remisión más prolongados entre los ataques, a diferencia de lo documentado en el síndrome de Majeed. Adicionalmente, esta condición no está directamente ligada a la anemia, aunque sí puede cursar con afecciones dermatológicas (tales como psoriasis, pustulosis palmoplantar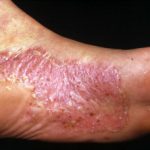 Pustulosis Palmoplantar: Causas, Síntomas y Condiciones Asociadas ¿Qué es la Pustulosis Palmoplantar? La pustulosis palmoplantar es un trastorno cutáneo crónico y poco frecuente, caracterizado por la aparición de pústulas estériles localizadas exclusivamente en las palmas de las manos y las plantas de los pies. También conocida formalmente como pustulosis palmaris et plantaris, esta condición a menudo se encuentra relacionada con la psoriasis, una afección dérmica más común. Una manifestación menos más o el síndrome de Sweet), afectando también las articulaciones y el tracto intestinal.
Pustulosis Palmoplantar: Causas, Síntomas y Condiciones Asociadas ¿Qué es la Pustulosis Palmoplantar? La pustulosis palmoplantar es un trastorno cutáneo crónico y poco frecuente, caracterizado por la aparición de pústulas estériles localizadas exclusivamente en las palmas de las manos y las plantas de los pies. También conocida formalmente como pustulosis palmaris et plantaris, esta condición a menudo se encuentra relacionada con la psoriasis, una afección dérmica más común. Una manifestación menos más o el síndrome de Sweet), afectando también las articulaciones y el tracto intestinal.
El diagnóstico riguroso del Síndrome de Majeed, que combina la evaluación genética con estudios de imagen avanzados como la RM y el análisis histopatológico, es crucial para instaurar un tratamiento apropiado y minimizar las complicaciones a largo plazo en el desarrollo del paciente.
Manejo Terapéutico Específico para el Síndrome de Majeed
El enfoque terapéutico se individualiza basándose en las manifestaciones clínicas predominantes que presente el paciente:
Tratamiento Dirigido a la Osteomielitis
- Se inicia con la administración de fármacos antiinflamatorios no esteroideos (AINE).
- Se pueden aplicar ciclos cortos controlados de corticosteroides sistémicos.
- Es vital integrar programas de fisioterapia para mantener tanto la función articular como la capacidad muscular.
- Se enfatiza la necesidad de evitar el reposo absoluto y prolongado en cama.
- Se ha reportado la ineficacia de la colchicinaPropiedades y Uso de la Colchicina en Dermatología La colchicina es un fármaco ancestral, derivado del azafrán de otoño, o *Colchicum autumnale* (una planta venenosa originaria de Europa). Históricamente, su aplicación principal ha sido el manejo de la gota. Aunque no está formalmente indicada ni aprobada para el tratamiento de afecciones dermatológicas, la colchicina se ha prescrito con éxito para ciertas patologías cutáneas debido a sus notables propiedades inmunológicas y más en los casos documentados de tres pacientes.
Manejo de la Anemia Asociada
- Monitoreo constante mediante recuentos sanguíneos periódicos.
- La consideración de soporte transfusional sanguíneo, si la anemia es severa.
- Evaluación para el procedimiento de esplenectomía en casos indicados.
Abordaje de las Afecciones Cutáneas
- Administración de corticoides orales administrados en cursos de corta duración.
Para futuras gestaciones, se puede considerar la implementación de pruebas prenatales, puesto que los hermanos de un niño afectado presentan una probabilidad del 25% de desarrollar el síndrome y un 50% de ser portadores de la mutación genética subyacente. Es indispensable aclarar que la viabilidad de estas pruebas depende de la identificación previa y precisa de la mutación en el niño ya diagnosticado. Asimismo, es un requisito obligatorio que todos los descendientes de un individuo con diagnóstico confirmado porten la alteración genética identificada.


