Wat is Sneddon? syndroom?
Het Sneddon-syndroom is zeldzaam, langzaam progressief, neurocutaan vasculopathie. Het wordt gekenmerkt door de combinatie van leven racemosa en terugkerend cerebrovasculair evenementen, zoals vergankelijk ischemisch aanvallen en beroertes.
Het Sneddon-syndroom wordt ook het Ehrmann-Sneddon-syndroom, livedo racemose en beroerte-syndroom, en livedo reticularis en beroerte-syndroom genoemd.
Wie krijgt het Sneddon-syndroom?
de incidentie van het Sneddon-syndroom bedraagt ongeveer vier per miljoen per jaar in de algemene bevolking. Het Sneddon-syndroom wordt meestal voor het eerst gediagnosticeerd bij vrouwen in de leeftijd van 20 tot 42 jaar. Er lijkt geen sprake te zijn van enige raciale variatie, hoewel de gerapporteerde aantallen klein zijn.
Het Sneddon-syndroom wordt vaak geassocieerd met auto immuun ziekten, zoals het antifosfolipidensyndroom en systemisch lupus erythematosus.
Wat veroorzaakt het Sneddon-syndroom?
Als antifosfolipide antilichaam markers aanwezig zijn in de 50-80% van patiënten met het Sneddon-syndroom, is het opgenomen in het klinische spectrum van primair antifosfolipidensyndroom.
Vasculair trombose en rechanneling van kleine en middelgrote bedrijven aderen in de huid en hersenen zijn het gevolg van coagulopathie of trombofilie, wat gepaard gaat met verminderde activiteitsniveaus eiwit C en proteïne S en verhoogde niveaus van factor VII.
Het Sneddon-syndroom kan worden geërfd als autosomaal dominante ziekte met variabele penetratie in zeldzaam familie gevallen. Verlies van functie mutaties zijn geïdentificeerd in cat eye-syndroom kandidaatgebied 1 (CECR1). chromosoom 22, dat codeert voor adenosinedeaminase 2 (ADA2).
Wat zijn de klinische kenmerken van het Sneddon-syndroom?
Het Sneddon-syndroom heeft dermatologische effecten en neurologisch demonstraties.
Dermatologische manifestaties
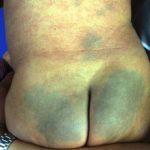
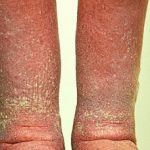
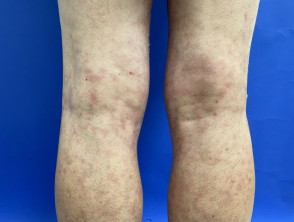
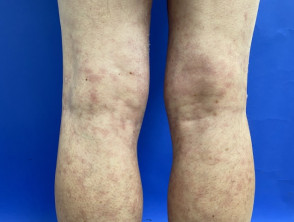
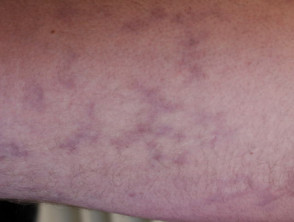
Livedo racemosa en acrocyanose
Livedo Racemosa
Livedo Racemosa
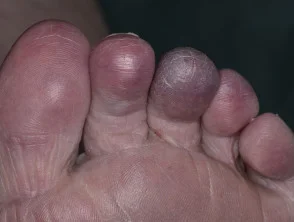
Acrocyanose
Het Sneddon-syndroom wordt gekenmerkt door livedo racemosa, die meer dan 10 jaar voorafgaat aan het begin van recidiverende beroertes. [1,2]. Het belang van livedo racemosa, dat vaak in de kindertijd begint, wordt pas in de jaren twintig en dertig onderkend nadat cerebrovasculaire gebeurtenissen zijn begonnen.
- Livedo racemosa is te wijten aan aanhoudend obstructie van perifere bloedstroom als gevolg van occlusie klein of middelgroot slagaders.
- Het verschijnt als een vertakkingspatroon in de vorm van een donker netwerk erythemateus of verkrachtig gebroken cirkels
- In tegenstelling tot livedo reticularis blijft de verkleuring van de huid onveranderd bij opwarming, hoewel deze prominenter aanwezig is tijdens blootstelling aan kou en zwangerschap.
- Livedo racemosa beïnvloedt aanvankelijk de bilspieren en de onderrug.
- Vooruitgang geleidelijk aan het betrekken van de dorsaal aspecten van de dijen en armen.
- Het heeft zelden invloed op het gezicht, de handen of de voeten.
- verwondingen zijn pijnloos en zijn niet geassocieerd met oedeem, ulceratieof jeuk.
Andere dermatologische manifestaties geassocieerd met het Sneddon-syndroom zijn onder meer:
- Acrocyanose
- Het fenomeen van Raynaud.
Neurologische manifestaties
De neurologische manifestaties die verband houden met het Sneddon-syndroom manifesteren zich doorgaans in drie fasen.
- Prodromaal symptomen kunnen omvatten Hoogtevrees, duizeligheid en hoofdpijn.
- In de tweede fase ervaren patiënten terugkerende episoden van beroerte of voorbijgaande ischemische aanvallen als gevolg van ischemie in de zones doorbloed door het midden of later cerebraal slagaders. Typische symptomen zijn onder meer afasie, gezichtsvelddefecten, hemiparese en sensorische stoornissen.
- De derde fase wordt gekenmerkt door aanzienlijke cognitieve stoornissen en beginnende dementie cumulatief meerdere herseneffecten hartaanvallen. Cognitief disfunctie, die het geheugen, de aandacht en de visueel-ruimtelijke domeinen beïnvloeden, en psychiatrisch Stoornissen, zoals depressie, worden gemeld bij ongeveer 77% van de patiënten met het Sneddon-syndroom.
Cerebrovasculaire complicaties van het Sneddon-syndroom zijn onder meer:
- Ischemische beroerte
- Hemorragisch beroerte (zeldzaam)
- toevallen
- Bewegingsstoornis (zeldzaam), zoals chorea of cerebellaire tremor.
Wat zijn de complicaties van het Sneddon-syndroom?
Cardiovasculaire complicaties van het Sneddon-syndroom zijn onder meer:
- Mitralis of aorta klep ziekte
- Libman-Sacks endocarditis
- Systolisch labiel hypertensie.
oogheelkundig Complicaties van het Sneddon-syndroom zijn onder meer:
- Optische schijf microaneurysma
- macula oedeem
- Netvlies slagader obstructie van de takken.
nier Complicaties zijn zeldzaam:
- Verminderde creatinineklaring
- Trombose van de nierslagader.
Hoe wordt het Sneddon-syndroom gediagnosticeerd?
De criteria voor de diagnose van het Sneddon-syndroom omvatten de presentatie van livedo racemosa met kenmerken histopathologisch bevindingen van de huid biopsie Y brandpunt neurologische tekorten. Een voorgeschiedenis van voorbijgaande ischemische aanvallen of beroerte of bewijs hiervan op beeldvorming kan de diagnose ondersteunen.
Patiënten met een vermoedelijke diagnose van het Sneddon-syndroom moeten bloedonderzoek ondergaan om auto-immuunziekten en coagulopathieën op te sporen. Magnetische resonantie, cardiovasculaire beoordeling en huidbiopsie.
Laesies geïdentificeerd door MRI zijn meestal klein, multifocaal en bevinden zich vaak in de diepe periventriculaire witte stof of pons. Bij maximaal 75% van de patiënten met het Sneddon-syndroom treden hersensymptomen op angiografie Het is abnormaal.
Bij een diepe huidbiopsie wordt histopathologie bewijst een neeopruiend trombotische vasculopathie waarbij sprake is van onderhuids klein en middelgroot dermaal slagaders. De gevoeligheid neemt toe met de hoeveelheid huid diep. biopsieën: 27%-gevoeligheid met één, 53% met twee en 80% met drie biopsieën.
Een grondig onderzoek is nodig bij patiënten met livedo racemosa of beroerte om andere ziekten in het differentieel uit te sluiten.
Classificatie
Het Sneddon-syndroom wordt geclassificeerd als antifosfolipiden-negatief of positief, afhankelijk van de aanwezigheid van een van de 3 antifosfolipiden. antistoffen: anticardiolipine, lupus anticoagulans en anti-bèta 2-glycoproteïne ik [2].
Het kan ook worden geclassificeerd als primair (idiopathisch) of secundair aan een geïdentificeerde trombofilie of auto-immuunziekte, zoals lupus erythematosus [1]. Het Sneddon-syndroom wordt af en toe ook geclassificeerd als vasculitis.
Welke is de differentiële diagnose voor het Sneddon-syndroom?
De differentiële diagnose van het Sneddon-syndroom kan het volgende omvatten:
- Aangeboren livedo reticularis
- Huid marmorata
- Ander hematologisch voorwaarde
- Antifosfolipidensyndroom
- cryoglobulinemie
- Polycytemie Vera
- Auto immuunziekte
- Polyarteritis nodosa
- Systemische vasculitis
- Livedoid vasculopathie
- Hartziekte resulterend in meerdere embolie
- Neoplasma
- lymfoom
- Leukemie
- Infectie
- Syfilis
- De ziekte van Lyme
- meningokokken ziekte
- Effecten van een medicijn
- Amantadine
- minocycline
- kinidine
- Divry van Bogaert-syndroom.
Wat is de behandeling voor het Sneddon-syndroom?
De behandeling is gebaseerd op anekdotische rapporten en omvat:
- Langdurige antistolling: bij patiënten die positief zijn voor antifosfolipide-antilichamen, wordt warfarine aanbevolen. Patiënten zonder antifosfolipide-antilichamen kunnen baat hebben bij bloedplaatjesaggregatieremmers
- Prostaglandine E1
- angiotensine conversie enzym (ACE) remmers
- Nifedipine: voor het fenomeen van Raynaud
- Immunosuppressivum medicijnen, zoals cyclofosfamide.
Aanbevolen levensstijlaanpassingen voor het Sneddon-syndroom zijn onder meer:
- Beëindiging van roken
- Beëindiging van oestrogeendie orale anticonceptiva bevatten
- Vermindering van beïnvloedbare cardiale risicofactoren.
Wat is het resultaat van het Sneddon-syndroom?
Het Sneddon-syndroom is een chronisch, een progressieve ziekte die vaak resulteert in cognitieve achteruitgang en vroegtijdige dementie als gevolg van het cumulatieve effect van meerdere herseninfarcten.