Wat is Waardenburg? syndroom?
Het Waardenburg-syndroom is zeldzaam genetisch stoornis die wordt gekenmerkt door doofheid en afwijkingen pigmentatie, Wat:
- witte pompadour
- Gehypopigmenteerd (bleke) plekken op de huid
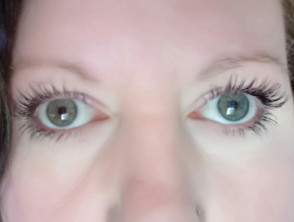
- Heterochromia iridis (verschillend gekleurde ogen)
Het Waardenburg-syndroom kan ook geassocieerd zijn met bewegingsapparaat defecten en het Hirschsprung-syndroom.
Het Waardenburgsyndroom is vernoemd naar Petrus Johannes Waardenburg, een Nederlander oogarts, die opmerkte dat heterochromia iridis vaak gepaard ging met doofheid.
Waardenburg-syndroom
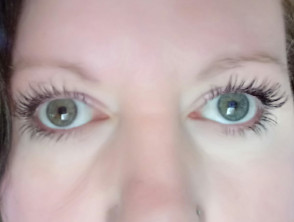
Heterochromia iridis
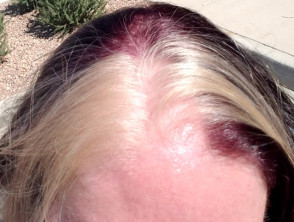
witte pompadour
Wie krijgt het Waardenburg-syndroom?
Het Waardenburg-syndroom kan erfelijk zijn of spontaan ontstaan. Het treft mannen en vrouwen in gelijke mate en kan alle rassen treffen.
Het heeft een populatiefrequentie van 1 op 42.000 en is verantwoordelijk voor 1-3% van alle gevallen van aangeboren doofheid.
Wat veroorzaakt het Waardenburg-syndroom?
Aangenomen wordt dat het Waardenburg-syndroom een neurocristopathie is. Het is het gevolg van afwijkingen neuraal kam differentiatie tijdens embryonaal ontwikkelen.
Verschillende gen mutaties kan het Waardenburg-syndroom veroorzaken, wat tot enkele verschillen in tekenen en symptomen leidt. Expressie en penetratie Ze zijn ook variabel.
- WS1 en WS3 worden veroorzaakt door mutaties in het PAX3-gen chromosoom 2q
- WS2A - MITF (geassocieerd met microftalmie transcriptie factor) gen, chromosoom 3p
- WS2B - chromosoom 1p
- WS2C - chromosoom 8p
- WS2D: SNAI2-gen, chromosoom 8q
- WS2E - SOX10-gen, chromosoom 22q
- WS4A – EDNRB-gen op 13q
- WS4B - Gen EDN3 op 20q
- WS4C - SOX10-gen (zoals voor WS2E)
Verschillende soorten mutatie kan het Waardenburg-syndroom veroorzaken: inserties, deleties, frameshifts, splitsingsveranderingen, missense of missense mutaties.
De meeste soorten Waardenburg-syndroom zijn dat wel autosomaal dominant, wat betekent dat slechts één aangetast gen aan een kind moet worden doorgegeven voordat het het syndroom krijgt. De overdracht van defecten in EDN3 of EDNRB is complexer. Dat zijn ze meestal autosomaal recessief, hoewel gevallen van autosomaal dominant Er zijn transmissies met onvolledige penetrantie beschreven.
Andere mutaties in sommige van de bovenstaande genen kan gerelateerde klinische syndromen veroorzaken, zoals het Tietz-syndroom (MITF-gen), piebaldisme (SNAI2-gen), PCWH (SOX10-gen) of ABCD-syndroom (EDNRB-gen).
Wat zijn de klinische kenmerken van het Waardenburg-syndroom?
De kenmerken zijn vanaf de geboorte aanwezig. Omdat het een zeldzame aandoening is en de klinische symptomen subtiel kunnen zijn, wordt de diagnose mogelijk pas op latere leeftijd gesteld.
Er zijn vier belangrijke klinische subtypes van het Waardenburg-syndroom geïdentificeerd.
Type 1
Type 1 is het meest voorkomende subtype van het Waardenburg-syndroom. Kenmerken omvatten:
- neurosensorisch doofheid
- Typische gezichtsstructuur, met dystopia canthorum (kant verplaatsing van de medium ooghoeken), brede neuswortel en synophrias (samenkomst van de wenkbrauwen ter hoogte van de ooghoeken). midden lijn)
- Hypogepigmenteerde plekken op de huid en haar
- Pigmentair afwijkingen van de ogen, zoals heterochromia iridis (verschillend gekleurde ogen), die compleet, gedeeltelijk of gedeeltelijk kunnen zijn segmentaal; isohypochromia iridis (lichtblauwe ogen); of abnormale pigmentatie van de fundus
Typ 2
Type 2 heeft vergelijkbare klinische kenmerken als het Waardenburg-syndroom type 1, maar de binnenste canthi zijn normaal.
typ 3
Type 3 (syndroom van Klein-Waardenburg) heeft ook vergelijkbare kenmerken als het syndroom van Waardenburg type 1, maar met afwijkingen aan het bewegingsapparaat, zoals spierhypoplasie, buiging contracturen of syndactylie (samengevoegd cijfers).
typ 4
Type 4 (Shah-Waardenburg-syndroom) heeft vergelijkbare kenmerken als het Waardenburg-syndroom type 2, maar met het Hirschsprung-syndroom (een aandoening die het gevolg is van de afwezigheid van zenuw spiercellen uit een deel of de gehele dikke darm).
Hoe wordt het Waardenburg-syndroom gediagnosticeerd?
De diagnose van het Waardenburg-syndroom is gebaseerd op klinische kenmerken. In 1992 ontwikkelde het Consortium Waardenburg grote en kleine criteria voor diagnose.
Belangrijkste criteria:
- Sensorineurale doofheid
- Pigmentachtige iris anomalie, als heterochromia iridis: compleet, gedeeltelijk of segmentaal; isohypochromie iridis; of pigmentafwijkingen van de fundus
- Afwijkingen in het haarpigment, zoals witte strepen, wenkbrauwen of wimpers
- Dystopia canthorum: laterale verplaatsing van de binnenste canthus
- Eerstegraads familielid met het Waardenburgsyndroom
kleine criteria
- Hypogepigmenteerde plekken op de huid
- Synofrys
- Brede neuswortel
- Alae nasi-hypoplasie
- Voortijdige veroudering van de hoofdhuid
Voor een klinische diagnose van het Waardenburgsyndroom type 1 zijn 2 hoofdcriteria of 1 hoofdcriterium en 2 nevencriteria vereist.
De W-index
De W-index kan worden berekend om te bepalen of dystopia canthorum aanwezig is.
- a = binnenste canthalafstand
- b = oogafstand
- c = buitenste canthalafstand
- X = (2a- (0,2119c + 3,909)) / c
- Y = (2a- (0,2479b + 3,909)) / b
- W = X + Y + a/b
- De W-index > 1,95 is indicatief voor dystopia canthorum.
Genetische sequencing van het PAX3-gen voor mutaties die het Waardenburg-syndroom veroorzaken, kan worden uitgevoerd als onderdeel van genetische counseling voor familieleden. Prenataal PAX3-mutatietesten zijn mogelijk door vlokkentest of vruchtwaterpunctie, maar het wordt zelden gedaan. Dit komt door de klinische variatie die wordt aangetroffen bij het Waardenburg-syndroom, aangezien de aanwezigheid van de mutatie niet aangeeft welke klinische kenmerken aanwezig zullen zijn of hoe ernstig deze zijn.
Wat is de behandeling voor het Waardenburg-syndroom?
Er bestaat geen directe behandeling voor het Waardenburg-syndroom zelf, en aangezien het een genetische ziekte is, is er geen genezing mogelijk. Genetische counseling kan nuttig zijn voor getroffen patiënten die een gezin willen stichten.
- Er moeten audiologische onderzoeken worden uitgevoerd bij kinderen met een vermoeden van het Waardenburg-syndroom om hun doofheid te beoordelen. Mogelijk zijn hoortoestellen of cochleaire implantaten nodig.
- Bij patiënten met het Hirschsprung-syndroom kan een operatie nodig zijn om het aangetaste deel van de darm te verwijderen.
- Hypogepigmenteerde plekken op de huid zijn meer gevoelig tegen schade door de zon, dus bescherming tegen de zon is belangrijk.
Wat is het resultaat van het Waardenburg-syndroom?
Het Waardenburg-syndroom is een chronisch conditie en kenmerken blijven een leven lang behouden. De levensverwachting is normaal.