Wat is Legius? syndroom?
Legius-syndroom is zeldzaam genetisch stoornis die voor het eerst werd beschreven in 2007 [1]. Ook gekend als neurofibromatose type 1-syndroom [2].
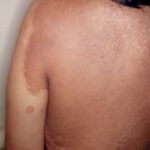
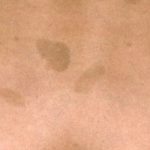
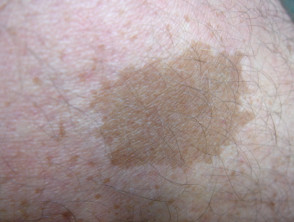
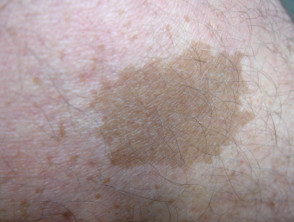
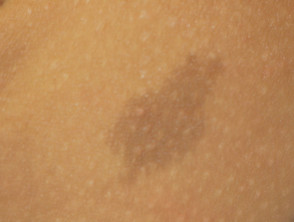
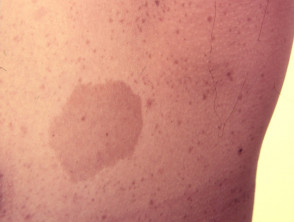
Legius-syndroom wordt klassiek gekenmerkt door meerdere macules, bekend als café-au-lait macules [3]. In tegenstelling tot type 1 neurofibromatose worden bij het syndroom van Legius geen tumoren gevonden.
Cafe-au-lait macules
café au lait macule
café au lait macule
café au lait macule
Wie krijgt het Legius-syndroom?
Legiussyndroom kan voorkomen bij iedereen die ten minste één biologische ouder heeft met genetisch bevestigd Legiussyndroom [2]. de overwicht van het Legius-syndroom is onbekend, maar kan hoger zijn dan verwacht vanwege een verkeerde diagnose van neurofibromatose type 1 [4].
Wat veroorzaakt het Legius-syndroom?
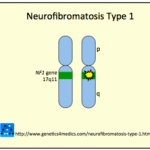
Het Legius-syndroom is een genetische aandoening die wordt geërfd in a autosomaal dominante manier die een impliceert LENTE1 gen mutatie in chromosoom 15q14. Deze mutatie resulteert in een abnormale functie van SPRED1 eiwit, die verantwoordelijk is voor het reguleren van de specifieke celsignaleringsroutes die betrokken zijn bij de proliferatie, differentiatieY apoptose. geen ander ziekteverwekker Er zijn varianten gevonden die het Legius-syndroom veroorzaken. [23].
Als een autosomaal dominant aandoening, kan het Legius-syndroom voorkomen bij iedereen met ten minste één biologische ouder met genetisch bevestigd Legius-syndroom. In zeldzame gevallen kan het ook voorkomen de nieuwe, maar deze gevallen zijn niet voldoende geëvalueerd om dit te bevestigen [2]. Elk kind met een ouder met het genetisch bevestigde Legius-syndroom heeft een 50%-risico om de aandoening te erven.
Wat zijn de klinische kenmerken van het Legius-syndroom?
De klinische kenmerken van het Legius-syndroom variëren aanzienlijk in aard en ernst van persoon tot persoon.
Bijna alle patiënten met het syndroom van Legius presenteren zich met meerdere café-au-lait macules [2,3]. Het aantal van deze macules neemt toe tijdens de kindertijd. [4].
ander gebruikelijk huid kenmerken kunnen zijn:
- Sproeten in de oksels en lies Regio's
- Lipomen [2-4].
Niet-huidkenmerken kunnen zijn:
- macrocefalie
- Ongebruikelijke gelaatstrekken, vergelijkbaar met het Noonan-syndroom
- Korte gestalte
- Pectus excavatum (verzonken borstbeen) of pectus carinatum (uitstekend borstbeen) [2–4].
Neuropsychiatrische kenmerken kunnen zijn:
- Leer moeilijkheden
- ontwikkelingsgericht vertragen
- Aandachtstekortstoornis met hyperactiviteit (ADHD) [2–4].
Verstandelijke beperkingen zijn over het algemeen minder ernstig bij patiënten met het syndroom van Legius in vergelijking met patiënten met neurofibromatose type 1 [5].
Het is belangrijk op te merken dat het Legius-syndroom niet veroorzaakt neurofibromen en Lisch knobbeltjes, die doorgaans worden gezien bij neurofibromatose type 1 [2,3].
Hoe wordt het Legius-syndroom gediagnosticeerd?
Het syndroom van Legius is alleen op klinische basis moeilijk te diagnosticeren, gezien de klinische presentatie van de huid die vergelijkbaar is met andere aandoeningen met meerdere café-du-lait maculae.
Wanneer het Legius-syndroom wordt vermoed, kunnen genetische tests worden gebruikt om de diagnose te bevestigen. Dit betrekt:
- Sequentie analyse LENTE1
- Deletie/duplicatie-analyse (als sequentie-analyse onopvallend is)
- Een multi-genenpanel voor LENTE1 en een ander genen interessant [2,3].
Verdachte bevindingen die genetische tests voor het Legius-syndroom kunnen rechtvaardigen, zijn onder meer:
- Cafe-au-lait macules zonder andere klinische kenmerken, suggestief voor neurofibromatose type 1
- Een vader met café-au-lait macules zonder klinische kenmerken, opnieuw suggestief voor neurofibromatose type 1 [2].
Welke is de differentiële diagnose voor het Legius-syndroom?
Het Legius-syndroom wordt vaak verkeerd gediagnosticeerd omdat het pigmentatie demonstraties lijken erg op die waargenomen bij andere syndromen met meerdere lentigines. De juiste diagnose is essentieel om de controle en het beheer op lange termijn te sturen.
Neurofibromatose type 1
- Neurofibromatose type 1 is de meest voorkomende verkeerde diagnose vanwege de vergelijkbare klinische diagnostische criteria [2-4].
- Neurofibromen, Lisch knobbeltjes, bot afwijkingenen optische gliomen komen niet voor bij het Legius-syndroom.
- Deze twee aandoeningen zijn moeilijk te onderscheiden bij jongere kinderen, omdat de karakteristieke tumoren die worden gezien bij neurofibromatose dat meestal niet doen. ontwikkelen tot later in het leven.
Noonan-syndroom met meerdere lentigines
- Noonan-syndroom met meerdere lentigines (voorheen bekend als LEOPARD-syndroom, omvat cardiovasculaire, oor- en genitale afwijkingen, die niet voorkomen bij het Legius-syndroom) [6].
Andere syndromen die worden gekenmerkt door meerdere lentigines zijn onder meer:
- autosomaal dominant café au lait macules
- McCune-Albright-syndroom.
Wat is de behandeling voor het Legius-syndroom?
Kinderen met het Legius-syndroom moeten regelmatig worden onderzocht en geëvalueerd om ontwikkelingsachterstanden, cognitieve achteruitgang en gedragsproblemen op te sporen. [2].
Behandeling van het Legius-syndroom is in de eerste plaats ondersteunend en moet zich richten op de specifieke problemen van de getroffen persoon.
Passend beheer, indien geïndiceerd, kan het volgende omvatten:
- Fysiotherapie, logopedie en/of ergotherapie
- Gedragsveranderingstherapie en medicamenteuze therapie (dwz voor ADHD) [2].
Wat is de uitkomst van het Legius-syndroom?
Patiënten met het syndroom van Legius hebben een goede voorspelling met goed probleemgestuurd management.
Een getroffen persoon moet rekening houden met het risico van genetische overdracht van 50% op elk kind.