¿Qué es la epidermólisis ampollosa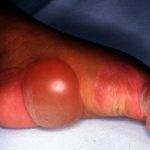 Epidermólisis Ampollosa: Un Trastorno Genético de la Piel La Epidermólisis Ampollosa (EB) es un complejo grupo de afecciones hereditarias caracterizadas por la propensión extrema de la piel y las membranas mucosas a formar ampollas y lesiones (lesiones). Estas vesículas cutáneas aparecen típicamente en zonas sujetas a fricción o mínimo trauma, como las manos y los pies. Sin embargo, en algunos subtipos genéticos, las ampollas pueden manifestarse internamente, afectando órganos como más con congénito ausencia de piel?
Epidermólisis Ampollosa: Un Trastorno Genético de la Piel La Epidermólisis Ampollosa (EB) es un complejo grupo de afecciones hereditarias caracterizadas por la propensión extrema de la piel y las membranas mucosas a formar ampollas y lesiones (lesiones). Estas vesículas cutáneas aparecen típicamente en zonas sujetas a fricción o mínimo trauma, como las manos y los pies. Sin embargo, en algunos subtipos genéticos, las ampollas pueden manifestarse internamente, afectando órganos como más con congénito ausencia de piel?
La epidermólisis ampollosa (EB) con ausencia congénita de piel se conocía anteriormente como Bart síndrome. También se le ha conocido como ‘tipo VI aplasia cutis congénita y epidermólisis ampollosa ».
La EB con ausencia congénita de piel fue descrita originalmente por Bruce Bart en 1966 después de observar una gran familia de seis generaciones con un simétrico ausencia congénita de piel en la parte inferior de las piernas, ampollas que afectan principalmente acral piel y a veces mucoso membranas, y uña distrofia [1]. En 1986, Ilona Frieden propuso una clasificación de aplasia cutis congénita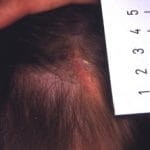 Definición y Características de la Aplasia Cutis Congénita La Aplasia Cutis Congénita (ACC) se define como la ausencia congénita de piel. La manifestación más habitual es un defecto localizado en el cuero cabelludo presente al momento del nacimiento. Es importante destacar que la aplasia cutis también forma parte integral de diversos síndromes genéticos asociados. Información clave sobre Aplasia Cutis Aplasia cutis Aplasia cutis Factores de Riesgo y Causas de la más en la que las condiciones se clasificaron en nueve grupos según la ubicación de lesiones y presencia de otras anomalías [2]. Según el sistema de clasificación de Friedan, el síndrome de Bart se clasificó como aplasia cutis congénita tipo VI por su combinación de un trastorno congénito. localizado ausencia de piel, epidermólisis ampollosa y distrofia ungueal [2].
Definición y Características de la Aplasia Cutis Congénita La Aplasia Cutis Congénita (ACC) se define como la ausencia congénita de piel. La manifestación más habitual es un defecto localizado en el cuero cabelludo presente al momento del nacimiento. Es importante destacar que la aplasia cutis también forma parte integral de diversos síndromes genéticos asociados. Información clave sobre Aplasia Cutis Aplasia cutis Aplasia cutis Factores de Riesgo y Causas de la más en la que las condiciones se clasificaron en nueve grupos según la ubicación de lesiones y presencia de otras anomalías [2]. Según el sistema de clasificación de Friedan, el síndrome de Bart se clasificó como aplasia cutis congénita tipo VI por su combinación de un trastorno congénito. localizado ausencia de piel, epidermólisis ampollosa y distrofia ungueal [2].
En 2014, un grupo de trabajo de expertos en EB sugirió una nueva clasificación de EB basada en molecular caracteristicas [3]. Sugirieron sustituir los nombres epónimos por términos descriptivos y recomendaron que el síndrome de Bart se conociera como EB con ausencia congénita de piel. [3].
¿Quién contrae epidermólisis ampollosa con ausencia congénita de piel?
La EB con ausencia congénita de piel es rara y su exacta incidencia es desconocido. La aplasia cutis congénita se observa en 1 a 2 de cada 10.000 nacimientos [4].
La EB con ausencia congénita de piel puede considerarse una forma rara de aplasia cutis congénita, pero se desconoce la proporción de aplasia cutis congénita que se puede clasificar como EB con ausencia congénita de piel.
¿Qué causa la epidermólisis ampollosa con ausencia congénita de piel?
La EB con ausencia congénita de piel es un familiar condición con un autosómico patrón dominante de herencia, pero también se han reconocido casos aislados [5].
los etiología y Patogénesis es complejo ya que la EB puede afectar diferentes membranas de la piel; Puede ser epidérmico (simplex), de unión o dérmico (distrófico). La familia original descrita por Bart tenía EB distrófica con cambios ultraestructurales en el anclaje fibrillas en el dermis. los genético anomalía en esa familia ha sido identificado como relacionado con el COL7A1 gene en cromosoma 3, que codifica para colágeno tipo VII [6,7].
Se cree que la ausencia congénita de piel es secundaria a la piel. fragilidad y en el útero trauma, explicando el simétrico distribución en la parte inferior de las piernas que pueden rozar, en lugar de como una falla para formar la piel [7].
¿Cuáles son las características clínicas de la epidermólisis ampollosa con ausencia congénita de piel?
La EB con ausencia congénita de piel es una tríada clínica que consta de:
- Ausencia congénita de piel en la parte inferior de las piernas.
- Cualquier tipo de EB en otra parte del cuerpo.
- Cambios en las uñas, como la ausencia congénita de uñas o distrofia ungueal.
Las áreas de piel ausente suelen ser simétricas y bilateral, por lo general involucrando medio o dorsal superficie de la distal miembros inferiores, incluida la parte superior de los pies. Son bruscamente demarcado, áreas rojas brillantes de ulceración [8].
En la familia original descrita por Bart, el fenotipo mostró cierta variabilidad, con no todos los miembros afectados mostrando todas las características de la tríada [7].
La EB con ausencia congénita de piel también puede estar asociada con otras anomalías, como:
- Atresia pilórica (obstrucción del píloro o la parte inferior del estómago)
- Oído rudimentario desarrollo
- Una nariz aplastada
- Una raíz nasal ancha (parte superior de la nariz)
- Ojos abiertos [5,8].
¿Cuáles son las complicaciones de la epidermólisis ampollosa con ausencia congénita de piel?
Las complicaciones de la EB con ausencia congénita de piel incluyen infección y hemorragia. Hipotermia, hipoglucemia, y los trastornos del equilibrio de líquidos pueden complicar grandes defectos [4,8]. Grave mucosa la participación ha causado una muerte prematura [7].
¿Cómo se diagnostica la epidermólisis ampollosa con ausencia congénita de piel?
La EB con ausencia congénita de piel es un diagnóstico clínico. Histológico La evaluación de la piel confirma el diagnóstico y categoriza el tipo de EB.
Biopsia ha revelado un subepidérmico ampolla con un inflamatorio infiltrado en la dermis en algunos casos [8]. En la serie de casos original de Bart, la división en la piel estaba debajo del lámina densa en electron microscopía, lo que indica una EB distrófica dérmica [7]. En otros casos, microscopio de electrones hallazgos de separación de la unión dérmico-epidérmica e interrupción de la basal láminas son similares a las observadas en EB de unión [9]. En varios casos, la división ha estado por encima del membrana basal indicando el tipo epidérmico simplex [7].
Cuál es el diagnóstico diferencial para la epidermólisis ampollosa con ausencia congénita de piel?
Los diagnósticos diferenciales de EB con ausencia congénita de piel se incluyen a continuación.
- Otras formas de aplasia cutis congénita: la aplasia cutis es una anomalía congénita poco común caracterizada por una ausencia de piel que ocurre con mayor frecuencia en el cuero cabelludo y la bóveda craneal. La profundidad de las lesiones puede variar desde superficial erosión a la ausencia de todas las capas de la piel. Hay nueve subtipos de aplasia cutis congénita; La EB con ausencia congénita de piel se ha clasificado como tipo VI.
- Otras formas de EB - EB es un grupo de trastornos hereditarios raros caracterizados por la formación de ampollas debido al aumento de piel y membrana mucosa fragilidad. Se han descrito cuatro tipos principales de EB según el nivel de piel escote a nivel ultraestructural. La EB se clasifica además en función de la herencia, los hallazgos clínicos y los defectos moleculares.
- Síndrome de Adams-Oliver```html Definición y Características del Síndrome de Adams-Oliver ¿Qué es el Síndrome de Adams-Oliver? El síndrome de Adams-Oliver es una enfermedad congénita poco frecuente, caracterizada por una serie de malformaciones notables en las extremidades, junto con un desarrollo anormal de la piel, especialmente en el cuero cabelludo. El síndrome de Adams-Oliver también recibe varias designaciones alternativas, tales como: • Ausencia de defectos de extremidades, cuero cabelludo y cráneo • Aplasia más: el síndrome de Adams-Oliver es una afección poco común caracterizada por aplasia cutis congénita del cuero cabelludo con transverso defectos de las extremidades y manchas de la piel.
¿Cuál es el tratamiento de la epidermólisis ampollosa con ausencia congénita de piel?
El tratamiento de la EB con ausencia congénita de piel suele ser conservador. El objetivo es acelerar la cicatrización de las áreas de piel afectadas, prevenir infecciones u otras complicaciones y reducir el riesgo de cicatrices.
Actual antibacteriano ungüento y los apósitos húmedos son los pilares del tratamiento [8]. Profiláctico sistémico no se recomiendan los antibióticosComprendiendo los Antibióticos: Definición, Historia y Clasificación Química Fundamentos de los Antibióticos: Definición y Alcance Los antibióticos son compuestos químicos esenciales diseñados para erradicar o inhibir el crecimiento de bacterias. Estrictamente, este término se refiere a los agentes antiinfecciosos orgánicos derivados de mohos o bacterias que son tóxicos para otros tipos de bacterias. No obstante, en el uso moderno y generalizado, el término "antibiótico" se ha ampliado para abarcar compuestos más. En ocasiones, puede ser necesaria una intervención quirúrgica con un injerto de piel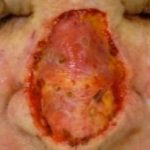 Conceptos Clave de los Injertos de Piel Un injerto de piel consiste en transferir una lámina de tejido cutáneo desde una zona del cuerpo hacia un área que presenta una pérdida significativa de piel. Esta intervención se vuelve necesaria frecuentemente después de la ablación quirúrgica de un cáncer o como secuela de lesiones graves, como quemaduras u otro trauma. A diferencia de un colgajo cutáneo, el injerto no conserva su más o una reparación con colgajo para un defecto grande [9].
Conceptos Clave de los Injertos de Piel Un injerto de piel consiste en transferir una lámina de tejido cutáneo desde una zona del cuerpo hacia un área que presenta una pérdida significativa de piel. Esta intervención se vuelve necesaria frecuentemente después de la ablación quirúrgica de un cáncer o como secuela de lesiones graves, como quemaduras u otro trauma. A diferencia de un colgajo cutáneo, el injerto no conserva su más o una reparación con colgajo para un defecto grande [9].
¿Cuál es el resultado de la epidermólisis ampollosa con ausencia congénita de piel?
En general, el pronóstico de EB con ausencia congénita de piel es bueno. La mayoría de las áreas de ausencia congénita de piel se curan en semanas o meses sin secuelas [7,10]. Sin embargo, cuando se volvió a visitar la familia original descrita por Bart, muchos adultos habían continuado desarrollar ampollas y milia y se observaron cicatrices leves en los sitios de formación de ampollas. Los sitios de la ausencia congénita de piel se habían curado con atrófico cicatrices sin pelo [7]. Las uñas no se recuperaron.


