¿Qué es el Síndrome de Angelman?
El síndrome de Angelman es un trastorno neurológico poco frecuente que afecta aproximadamente a 1 de cada 15,000 nacimientos. Históricamente, se confundía con otras condiciones como la parálisis cerebral o el autismo, debido a su complejo formación de síntomas distintivos.
Este síndrome fue nombrado en honor al Dr. Harry Angelman, quien describió la afección por primera vez en 1965.
¿A quién afecta el Síndrome de Angelman?
La naturaleza del síndrome de Angelman es predominantemente esporádica:
- La gran mayoría de las personas que padecen el síndrome de Angelman no tienen antecedentes familiares conocidos de la afección.
- En un porcentaje reducido de casos, la condición puede ser hereditaria de uno de los progenitores.
- La literatura médica no señala predilecciones específicas basadas en raza o sexo.
¿Cuál es la causa genética del Síndrome de Angelman?
El síndrome de Angelman es de origen genético. Las alteraciones genéticas pueden surgir de manera aleatoria, incluso en ausencia de antecedentes familiares de este trastorno.
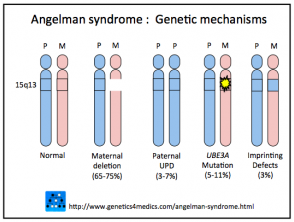
- Se origina por la pérdida de expresión de la copia materna del gen UBE3A, que se localiza en el cromosoma 15q11.2-q13OCA2.
- La deleción cromosómica es responsable entre el 65% y el 75% de los casos.
- Las mutaciones del gen materno afectan entre el 5% y el 11% de los pacientes.
- La disomía uniparental paterna (pUPD, donde ambas copias cromosómicas provienen del padre) incide en el 3% al 7% de los casos.
- Un defecto de impronta materna es la causa en aproximadamente el 3% de los diagnósticos.
El síndrome de Prader-Willi es un trastorno clínicamente diferenciado que surge de un defecto de origen paterno mapeado en el mismo sector cromosómico que el síndrome de Angelman.
Imágenes sobre la Genética del Síndrome de Angelman
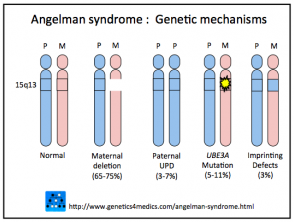
Genética del síndrome de Angelman
* Imagen cortesía de Genetics 4 Medics
Características Clínicas del Síndrome de Angelman
Cutáneas, las manifestaciones clínicas del síndrome de Angelman a menudo incluyen:
- Piel hipopigmentada.
- Albinismo```html Entendiendo el Albinismo: Tipos, Clasificación y Herencia Genética El albinismo es una condición congénita caracterizada por la escasa o nula producción de melanina, el pigmento esencial que otorga color a los ojos, la piel y el cabello. Esta ausencia de pigmentación hace que las personas afectadas presenten características físicas distintas a las de sus familiares sin esta condición: piel muy clara, altamente susceptible a las quemaduras solares, cabello blanco más ocular y cutáneo.
- Deficiencia en el pigmento de la coroides.
- Melanización incompleta de los melanosomas.
- Coloración azul claro en el iris.
- Reducción del pigmento retiniano.
- Baja actividad de la cabello bulbo tirosinasa.
Además de las características cutáneas, las manifestaciones no cutáneas del síndrome de Angelman abarcan convulsiones, retrasos de desarrollo, lenguaje limitado o ausente, alteraciones en la movilidad, una sonrisa frecuentemente aumentada, personalidad alegre y excitable, aleteo de las manos, patrones de sueño anómalos y microcefalia.
Tratamiento para el Síndrome de Angelman
Actualmente no existe una cura para el síndrome de Angelman. La atención requerida es continua y el tratamiento se enfoca en la gestión sintomática. Este manejo puede incluir:
- Medicamentos anticonvulsivos (anti-convulsiones), como el valproato de sodio, administrado solo o en combinación con clonazepam u otra benzodiazepina.
- Terapia de comunicación para instruir a los niños en el uso del lenguaje de signos o sistemas de imágenes.
- Terapia conductual diseñada para asistir en el manejo de la hiperactividad y los períodos cortos de atención.
Pronóstico del Síndrome de Angelman
A pesar de las múltiples limitaciones funcionales asociadas, la esperanza de vida para los pacientes con síndrome de Angelman suele ser normal. Conforme envejecen, es común que los pacientes experimenten una reducción en la excitabilidad y superen las anormalidades del ciclo del sueño.


