Comprendiendo las Porfirias: Definición y Tipos
¿Qué Son las Porfirias? Entendiendo la Vía del Hemo
Las porfirias constituyen un grupo de enfermedades de naturaleza genética caracterizadas por deficiencias enzimáticas críticas dentro de la vía de biosíntesis del hemo. El hemo, que es el componente de la hemoglobina responsable del color rojo de la sangre, requiere una secuencia precisa de reacciones catalizadas por enzimas específicas. Una deficiencia enzimática conduce a la acumulación de precursores químicos en la sangre y diversos órganos.
Dependiendo de la enzima afectada, los precursores pueden desencadenar ataques **agudos** en las porfirias hepáticas (**agudo**), o provocar fotosensibilidad Entendiendo la Fotosensibilidad: Definición y Clasificación Médica La fotosensibilidad abarca diversas afecciones, síntomas y enfermedades que son desencadenadas o se exacerban significativamente por la exposición a la radiación solar. • Una reacción cutánea provocada por la fotosensibilidad se denomina fotodermatosis (plural: fotodermatosis). • Cuando esta erupción presenta características eccematosas, se clasifica específicamente como una fotodermatitis. • Una sustancia química o un medicamento que induce fotosensibilidad es conocido como fotosensibilizador. • más y severas lesiones cutáneas en las porfirias **cutáneas**.
Entendiendo la Fotosensibilidad: Definición y Clasificación Médica La fotosensibilidad abarca diversas afecciones, síntomas y enfermedades que son desencadenadas o se exacerban significativamente por la exposición a la radiación solar. • Una reacción cutánea provocada por la fotosensibilidad se denomina fotodermatosis (plural: fotodermatosis). • Cuando esta erupción presenta características eccematosas, se clasifica específicamente como una fotodermatitis. • Una sustancia química o un medicamento que induce fotosensibilidad es conocido como fotosensibilizador. • más y severas lesiones cutáneas en las porfirias **cutáneas**.
¿Qué es la Protoporfiria Eritropoyética (EPP)?
La Protoporfiria Eritropoyética (EPP) se clasifica dentro del espectro de las porfirias cutáneas. Esta condición se origina por una deficiencia hereditaria de la enzima ferroquelatasa. La actividad reducida de esta enzima provoca la acumulación de protoporfirina, principalmente en la piel, lo que se manifiesta como una sensibilidad extrema a la luz solar.
Es importante notar que, en raras ocasiones, niveles excesivamente altos de protoporfirina pueden evolucionar hacia el desarrollo de enfermedad hepática.
¿Quién y Cuándo Desarrolla la Protoporfiria Eritropoyética?
La Protoporfiria Eritropoyética (EPP) es la segunda porfiria cutánea más frecuente, afectando aproximadamente a 1 de cada 100,000 individuos en la población general. Aunque se hereda en la inmensa mayoría de los casos, muchos afectados no presentan historial familiar debido a la naturaleza variable de su transmisión.
El riesgo de estar desarrollando EPP se estima en 1 de cada 10 descendientes cuando uno de los padres está afectado. La enfermedad afecta por igual a hombres y mujeres. Habitualmente, los primeros indicios sintomáticos se manifiestan durante la infancia temprana o el primer período de vida.
¿Qué Causa la Protoporfiria Eritropoyética? Bases Genéticas
Se postula que la EPP resulta de una pérdida compuesta de función hereditaria, específicamente una mutación en el gene responsable de codificar la ferroquelatasa (FECH; OMIM 612386), ubicado en el cromosoma 18q21. Típicamente, esto implica la presencia de una mutación en un alelo y una segunda mutación de baja expresión en el alelo complementario.
En circunstancias menos frecuentes, la causa puede ser una ganancia de función mutacional en la ALA sintasa (ALAS) -2 (herencia ligada al cromosoma X dominante). Por lo tanto, el patrón de herencia puede ser autosómico recesivo o dominante autosómico, aunque este último exhibe una penetrancia incompleta (reportada en el 96% de los pacientes en el Reino Unido).
Existen reportes extremadamente raros donde la EPP ha sido precipitada por condiciones como la mielodisplasia o la leucemia mieloide, vinculadas a inestabilidad cromosómica asociada, incluida la pérdida del cromosoma 18.
Manifestaciones Clínicas de la Protoporfiria Eritropoyética
Síntomas y Signos Cutáneos Característicos de la EPP
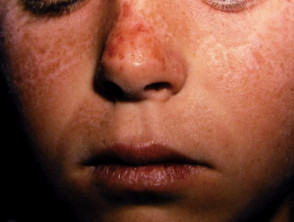
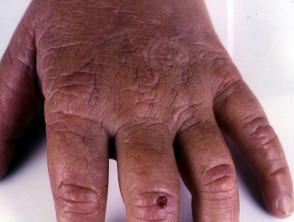
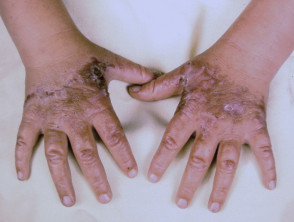
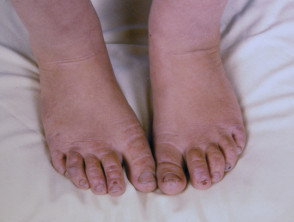
La EPP se manifiesta primariamente a través de dolor intenso en la piel tras la exposición a la luz solar. Esta reacción afecta con mayor frecuencia las superficies dorsales de las manos y los pies, así como la cara y las orejas. El dolor puede ser lo suficientemente severo como para persistir durante varios días después del cese de la exposición solar, y es posible que no exista evidencia visual inmediata de la lesión.
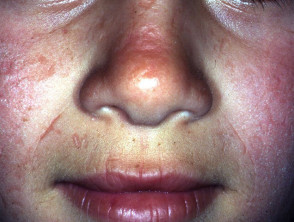
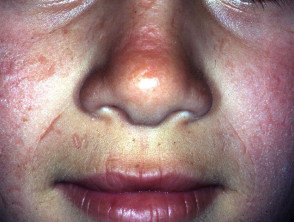
La exposición prolongada puede inducir enrojecimiento Componentes y Diagnóstico del Enrojecimiento Facial El enrojecimiento, conocido médicamente como rubor, se produce cuando los vasos sanguíneos superficiales de la piel se dilatan. Si esta dilatación es mediada por la actividad de los nervios que inervan dichos vasos, frecuentemente se acompaña de sudoración. Por otro lado, los agentes que actúan directamente sobre la pared vascular provocan típicamente un rubor seco, sin transpiración. Imágenes Ilustrativas del Enrojecimiento Enrojecimiento inducido por más e hinchazón. Con menor frecuencia, se pueden observar ampollas y la formación de costras.
Componentes y Diagnóstico del Enrojecimiento Facial El enrojecimiento, conocido médicamente como rubor, se produce cuando los vasos sanguíneos superficiales de la piel se dilatan. Si esta dilatación es mediada por la actividad de los nervios que inervan dichos vasos, frecuentemente se acompaña de sudoración. Por otro lado, los agentes que actúan directamente sobre la pared vascular provocan típicamente un rubor seco, sin transpiración. Imágenes Ilustrativas del Enrojecimiento Enrojecimiento inducido por más e hinchazón. Con menor frecuencia, se pueden observar ampollas y la formación de costras.
- La EPP puede
- Puede ser compleja de diagnosticar durante la infancia, dado que los bebés reaccionan al sol o al estar en el automóvil con llanto.
- Los síntomas suelen acentuarse durante el verano y en contextos de alta exposición solar.
- Con el paso de los años, la piel del dorso de las manos y las mejillas puede mostrar un ligero engrosamiento y presentar cicatrices sutiles con apariencia de hoyuelos.
- La intensidad de la reacción varía considerablemente: desde formas leves, que requieren varias horas de sol para causar dolor, hasta casos severos donde el dolor aparece en cuestión de segundos.
- En la mayor parte de los diagnósticos, las alteraciones cutáneas visibles son mínimas.
Protoporfiria Eritropoyética y Manifestaciones Clínicas
Protoporfiria eritropoyética
Protoporfiria eritropoyética
Protoporfiria eritropoyética
Protoporfiria eritropoyética
Protoporfiria eritropoyética
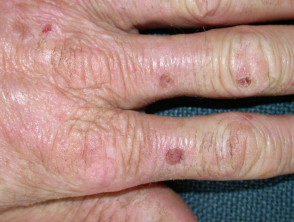
Protoporfiria eritropoyética
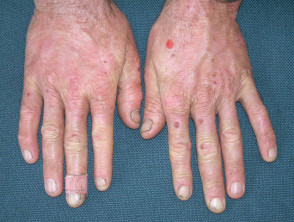
Protoporfiria eritropoyética
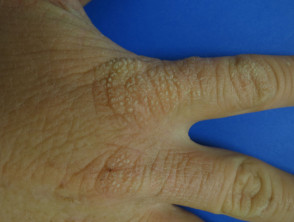
Protoporfiria eritropoyética
Impacto Hepático en la Protoporfiria Eritropoyética
Los pacientes con Protoporfiria Eritropoyética (EPP) que experimentan afectación hepática suelen mostrar resultados ligeramente alterados en los análisis de función hepática. Aproximadamente el 10% de los casos pueden desarrollar patología hepática avanzada. Esta progresión se manifiesta con malestar general, dolor localizado en el cuadrante superior derecho del abdomen, y posterior desarrollo de ictericia, pudiendo culminar en insuficiencia hepática (lo cual intensifica aún más la sensibilidad a la luz solar).
Además, la formación de cálculos biliares resulta ser un hallazgo frecuente en las personas diagnosticadas con EPP.
Diagnóstico y Tratamiento de la Protoporfiria Eritropoyética (PPE)
El proceso de diagnóstico de la Protoporfiria Eritropoyética (PPE) generalmente se inicia durante la infancia. Para confirmar la afección, es crucial enviar muestras de sangre para el análisis de porfirina, las cuales deben ser recolectadas en tubos protegidos rigurosamente de la exposición lumínica mediante papel de aluminio.
Métodos para el Diagnóstico de la Protoporfiria Eritropoyética
El diagnóstico de la PPE se apoya en varias pruebas clave, destinadas a identificar la acumulación anormal de protoporfirinas:
- Mediante microscopía de fluorescencia bajo luz ultravioleta, se puede observar la emisión de fluorescencia característica en los glóbulos rojos (eritrocitos) del paciente.
- El hallazgo bioquímico fundamental es la marcada elevación de la protoporfirina encontrada en los glóbulos rojos.
- Se pueden efectuar pruebas genéticas para detectar mutaciones específicas en el gen de la ferroquelatasa (FECH).
- Una biopsia cutánea es raramente necesaria dado que la piel suele presentar un aspecto normal; no obstante, la PPE revela ciertas particularidades en su histopatología.
Tras establecer el diagnóstico definitivo de PPE, es indispensable realizar un seguimiento periódico de la función hepática e incorporar estudios de imagen hepática de manera intermitente.
Opciones de Tratamiento para la Protoporfiria Eritropoyética
Actualmente, no existe una cura definitiva para la Protoporfiria Eritropoyética. La principal dificultad a largo plazo radica en el manejo de la fotosensibilidad crónica que afecta a los pacientes de por vida.
Cuando el dolor fotosensible ya se ha manifestado, su alivio puede ser complejo. La mayoría de los afectados encuentran consuelo sumergiendo las áreas afectadas en agua fría o aplicando compresas de hielo. En estos casos, el uso de cremas anestésico actuales puede ser beneficioso.
Para minimizar la intensidad del dolor, las estrategias de prevención son esenciales:
- Evitar cualquier exposición innecesaria a la luz solar directa y utilizar vestimenta protectora junto con sombreros de ala ancha.
- Se recomienda polarizar o tintar las ventanillas de los vehículos para reducir la penetración de luz.
- Otras fuentes de iluminación, como las luces halógenas y fluorescentes, también tienen el potencial de desencadenar síntomas.
- Es vital proteger la piel si se requiere exposición a lámparas durante procedimientos quirúrgicos.
- El empleo de protectores solares resulta útil, en particular aquellas formulaciones que contienen dióxido de titanio o bien óxido de zinc, sustancias que reflejan la luz visible.
- La suplementación con vitamina DGuía Esencial de la Vitamina D: Formas Bioquímicas, Síntesis y Exposición Solar Óptima La vitamina D abarca una familia de vitaminas liposolubles que existen en diferentes estructuras químicas. Comprender estas variantes es fundamental para entender su función integral en la fisiología humana. • La Vitamina D2 (ergocalciferol o calciferol) se origina a partir de la provitamina ergosterol, presente inactiva en los vegetales, tras ser activada por la radiación ultravioleta (UVG. más es una medida apropiada solo para aquellos pacientes que logran evitar rigurosamente cualquier contacto con la luz solar.
Además, restringir el consumo de alcohol es crucial para mitigar el riesgo de desarrollar complicaciones hepáticas graves e insuficiencia hepática.
La evaluación de tratamientos efectivos para la PPE ha presentado desafíos debido a que una terapia eficaz debe demostrar capacidad para reducir el dolor y, al mismo tiempo, incrementar el tiempo que el paciente puede pasar al aire libre sin experimentar molestias.
- La fototerapiaLa fototerapia constituye el uso estratégico de determinados tipos de radiación electromagnética con el fin de tratar diversas afecciones dermatológicas. Esta técnica emplea longitudes de onda específicas para modular la respuesta biológica de la piel. Visite las páginas de DermNet NZ sobre fototerapia para obtener información exhaustiva. Los métodos y aplicaciones principales de la fototerapia incluyen: • Tratamiento del acné mediado por luz azul. • Terapia con radiación ultravioleta B más utilizando luz UVB de banda estrecha ha demostrado no provocar el dolor asociado a la EPP. Este tratamiento se administra tres veces por semana durante un ciclo de seis semanas en primavera. El objetivo es aumentar el contenido de melanina (induciendo un bronceado
 Comprendiendo las Quemaduras Solares: Causas y Riesgos Definición y Mecanismos de las Quemaduras Solares Las quemaduras solares se manifiestan como eritema (enrojecimiento) e hinchazón o edema, resultantes de una exposición excesiva a la radiación solar, específicamente la radiación ultravioleta (UV) emitida por el sol. Si bien la exposición solar es la causa más común, también pueden originarse por otras fuentes de luz UV, como las lámparas solares o los salones más) y promover el engrosamiento dérmico para ofrecer una barrera protectora natural contra el sol.
Comprendiendo las Quemaduras Solares: Causas y Riesgos Definición y Mecanismos de las Quemaduras Solares Las quemaduras solares se manifiestan como eritema (enrojecimiento) e hinchazón o edema, resultantes de una exposición excesiva a la radiación solar, específicamente la radiación ultravioleta (UV) emitida por el sol. Si bien la exposición solar es la causa más común, también pueden originarse por otras fuentes de luz UV, como las lámparas solares o los salones más) y promover el engrosamiento dérmico para ofrecer una barrera protectora natural contra el sol. - Se han explorado antioxidantes orales como N-acetilcisteína, betacaroteno, extracto de Polypodium leucotomos y warfarina; sin embargo, la evidencia concluyente de su efectividad sigue siendo limitada.
- Debe evitarse la suplementación con hierro, a menos que exista una deficiencia severa diagnosticada, dado que el hierro puede intensificar la fotosensibilidad característica de la EPP.
- AfamelanotidaComprendiendo la Afamelanotida: Mecanismo y Aplicaciones Dermatológicas La afamelanotida (comercializada como SCENESSE® por Clinuvel Pharmaceuticals) es reconocida como un potente análogo de la hormona estimulante de los melanocitos alfa ($alphatext{MSH}$). Este compuesto sintético, conocido previamente como Melanotan I, actúa estimulando la biosíntesis de eumelanina en la piel, induciendo así un efecto de bronceado protector. Este péptido melanotrópico se administra mediante inyección. La eumelanina constituye la forma principal del pigmento de más, una hormona melanocito estimulante administrada mediante implantación subcutáneo, ha reportado beneficios clínicos significativos y un perfil de seguridad aceptable en casos de EPP. Este medicamento cuenta con aprobación como medicamento huérfano tanto en Estados Unidos como por la Agencia Europea de Medicamentos.
- Los pacientes con EPP que simultáneamente presentan enfermedad hepática requieren una atención médica altamente especializada y, en algunos casos, pueden necesitar un trasplante hepático.
Es importante notar que otra condición relacionada, la porfiria eritropoyética congénita (más grave), ahora puede ser curada mediante la transfusión de células madre, lo cual ofrece esperanza para el futuro del campo. No obstante, para la EPP, una cura terapéutica aún no está disponible.


