Wat is auto immuun polyglandulair syndroom Type 1?
Auto-immuun polyglandulair syndroom type 1 (APS1) is een auto-immuunziekte die resulteert in het falen van meerdere endocrien klieren. Ook bekend als auto-immuun polyendocrien syndroom type 1, polyendocrinopathie-candidiasis–ectodermaal dystrofie (APECED), Whitaker-syndroom en candidiasis-hypoparathyreoïdie-Addison-syndroom, naast de vele andere namen.
APS1 werd voor het eerst beschreven door Dr. Thomas Addison in de 19e eeuw.
Wie krijgt auto-immuun polyglandulair syndroom type 1?
APS1 is erfelijk en vrouwen en meisjes hebben iets meer kans dan mannen en jongens ontwikkelen syndroom. Het komt vaker voor in bepaalde etnische populaties vanwege bloedverwantschap of de groepering van afstammelingen van een gemeenschappelijke familiestichter. Het is meer overheersend in:
- Iraanse joden (1 op 9.000)
- Sardiniërs (1 op 14.400)
- Finnen (1 op 25.000).
APS1 komt niet vaak voor in andere populaties.
Wat veroorzaakt auto-immuun polyglandulair syndroom type 1?
APS1 wordt veroorzaakt door gen mutaties in het auto-immuun regulerende gen, LUCHT, in chromosoom 21q22. Het is geërfd in een autosomaal recessief patroon (er moeten twee kopieën van een abnormaal gen aanwezig zijn om het syndroom te ontwikkelen). Deze genetische mutaties leiden tot auto-antilichamen en oorzaak chronisch opruiend cel infiltranten in de aangetaste organen.
Wat zijn de klinische kenmerken van auto-immuun polyglandulair syndroom type 1?
Symptomen komen het vaakst voor bij kinderen van 3 tot 5 jaar, waarbij de meeste APS1-gevallen in de vroege adolescentie verschijnen en alle gevallen tegen de tijd dat iemand begin dertig is.
APS1 is gebaseerd op drie belangrijke klinische kenmerken:
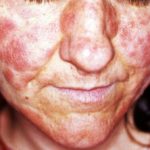
- mucocutaan candidiasis die de huid aantast en slijm- membranen
- Hypoparathyreoïdie, die gevoelloosheid en tintelingen in het gezicht en ledematen, spierpijn, zwakte en vermoeidheid veroorzaakt als gevolg van lage niveaus van circulerend calcium.
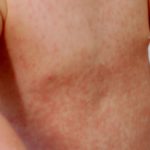
- De ziekte van Addison, een falen van de bijnieren, die gepaard gaat met huidveranderingen. pigmentatie, verlies van eetlust en gewichtsverlies, vermoeidheid, lage bloeddruk en vermoeidheid.
Hoewel minder vaak voorkomend, kunnen andere mogelijke kenmerken van dit syndroom zijn:
- hypogonadotroop hypogonadisme
- schadelijk Bloedarmoede
- chronisch actief hepatitis
- Asplenie
- Keratoconjunctivitis
- interstitieel nefritis
- diabetes mellitus type 1
- cholelithiase
- alopecia areata
- Malabsorptie
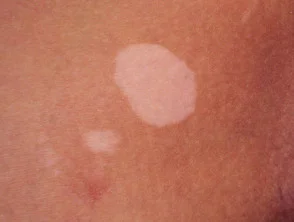
- vitiligo
Huidsymptomen van APS type 1
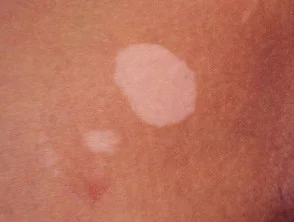
vitiligo
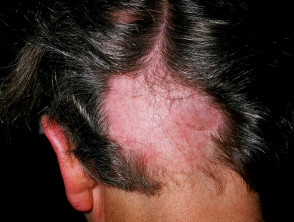
alopecia areata
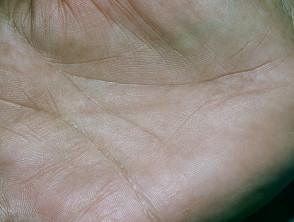
de ziekte van Addison
Hoe wordt auto-immuun polyglandulair syndroom type 1 gediagnosticeerd?
Als een persoon aanwijzingen heeft voor meer dan één endocriene deficiëntie, kunnen verdere tests worden uitgevoerd om auto-immuun polyglandulair syndroom type 1 (APS1) te bevestigen, waaronder:
- EEN serum auto-immuun scherm
- Beëindig orgaanfunctietesten.
Andere mogelijke bloedonderzoeken kunnen tests voor zijn testosteron, oestradiol, follikel-stimulerend hormoon (FSH), luteïniserend hormoon (LH), prolactine, adrenocorticotroop hormoon (ACTH), plasma renine-activiteit, elektrolytniveaus en volledig bloedbeeld.
Wattenstaafjes en huidafkrabsels kunnen ook worden genomen om te detecteren Candida albicans.
Hoe wordt auto-immuun polyglandulair syndroom type 1 behandeld?
De behandeling van APS1 hangt af van de specifieke kenmerken.
- Mucocutane candidiasis wordt behandeld met orale antischimmelmiddelen, zoals fluconazol of itraconazol.
- Hypoparathyreoïdie wordt behandeld met een combinatie van oraal calcium en vitamine D (meestal calcitriol).
- De ziekte van Addison wordt behandeld met corticosteroïden en mineralosteroïden. Bijnier klier transplantaties kunnen ook geïndiceerd zijn.
Wat is de uitkomst van auto-immuun polyglandulair syndroom type 1?
de voorspelling voor APS1 is het variabel. De overlevingspercentages zijn sinds de jaren zeventig sterk verbeterd.