Che cos'è autoimmune polighiandolare sindrome Tipo 1?
La sindrome polighiandolare autoimmune di tipo 1 (APS1) è una condizione autoimmune che provoca il fallimento di più endocrino ghiandole. Conosciuta anche come sindrome poliendocrina autoimmune di tipo 1, poliendocrinopatia-candidosi–ectodermico distrofia (APECED), sindrome di Whitaker e sindrome da candidosi-ipoparatiroidismo-morbo di Addison, tra i suoi molti altri nomi.
APS1 è stato descritto per la prima volta dal Dr. Thomas Addison nel 19° secolo.
Chi ha la sindrome autoimmune polighiandolare di tipo 1?
L'APS1 è ereditato e le donne e le ragazze hanno una probabilità leggermente maggiore rispetto agli uomini e ai ragazzi sviluppare sindrome. Si verifica più frequentemente in particolari popolazioni etniche a causa di consanguineità o il raggruppamento di discendenti di un comune capostipite. È più predominante su:
- Ebrei iraniani (1 su 9.000)
- Sardi (1 su 14.400)
- Finlandesi (1 su 25.000).
APS1 è raro in altre popolazioni.
Quali sono le cause della sindrome polighiandolare autoimmune di tipo 1?
APS1 è causato da gene mutazioni nel gene regolatore autoimmune, ARIA, in cromosoma 21q22. Si eredita in a autosomica pattern recessivo (per lo sviluppo della sindrome devono essere presenti due copie di un gene anomalo). Queste mutazioni genetiche portano a autoanticorpi e causa cronica infiammatorio cellula infiltrati negli organi colpiti.
Quali sono le caratteristiche cliniche della sindrome polighiandolare autoimmune di tipo 1?
I sintomi compaiono più spesso nei bambini di età compresa tra 3 e 5 anni, con la maggior parte dei casi di APS1 che compaiono nella prima adolescenza e tutti i casi quando una persona ha circa 30 anni.
APS1 si basa su tre caratteristiche cliniche principali:
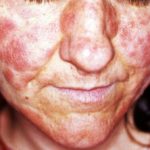
- Mucocutanea candidosi che colpisce la pelle e mucoso membrane
- Ipoparatiroidismo, che provoca intorpidimento e formicolio al viso e alle estremità, crampi muscolari e dolore, debolezza e affaticamento dovuti ai bassi livelli di calcio circolante.
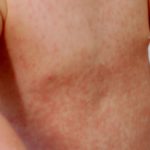
- Morbo di Addison, un'insufficienza delle ghiandole surrenali, che si presenta con alterazioni della pelle. pigmentazione, perdita di appetito e perdita di peso, affaticamento, pressione sanguigna bassa e affaticamento.
Sebbene meno comuni, altre possibili caratteristiche di questa sindrome possono includere:
- ipogonadotropo ipogonadismo
- Pernicioso anemia
- attivo cronico epatite
- asplenia
- Cheratocongiuntivite
- Interstitial nefrite
- Diabete mellito di tipo 1
- Colelitiasi
- Alopecia areata
- Malassorbimento
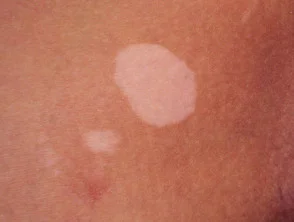
- Vitiligine
Segni cutanei di tipo APS 1
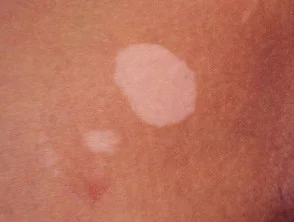
Vitiligine
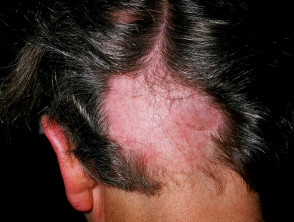
Alopecia areata
morbo di Addison
Come viene diagnosticata la sindrome polighiandolare autoimmune di tipo 1?
Se un individuo ha evidenza di più di un deficit endocrino, possono essere eseguiti ulteriori test per confermare la sindrome polighiandolare autoimmune di tipo 1 (APS1), tra cui:
- UN siero schermo autoimmune
- Terminare i test di funzionalità d'organo.
Altri possibili esami del sangue possono includere test per testosterone, estradiolo, follicolo-ormone stimolante (FSH), ormone luteinizzante (LH), prolattina, ormone adrenocorticotropo (ACTH), plasma attività della renina, livelli di elettroliti ed emocromo completo.
Per rilevare anche tamponi e raschiati cutanei candida albicans.
Come viene trattata la sindrome polighiandolare autoimmune di tipo 1?
Il trattamento di APS1 dipenderà dalle sue caratteristiche specifiche.
- La candidosi mucocutanea viene trattata con agenti antimicotici orali, come fluconazolo o itraconazolo.
- L'ipoparatiroidismo viene trattato con una combinazione di calcio orale e vitamina D (di solito calcitriolo).
- Il morbo di Addison è trattato con corticosteroidi e mineralosteroidi. Surrenale ghiandola possono essere indicati anche i trapianti.
Qual è l'esito della sindrome polighiandolare autoimmune di tipo 1?
il previsione per APS1 è variabile. I tassi di sopravvivenza sono migliorati molto dagli anni '70.