¿Qué es cobimetinib?
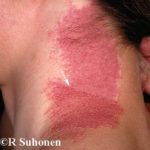
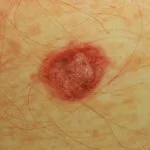
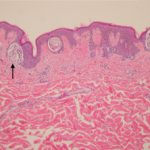
Cobimetinib es un medicamento recetado que se usa para tratar melanoma que se ha diseminado a otras partes del cuerpo, no se puede extirpar mediante cirugía y tiene una anomalía BRAF gene. No debe usarse para tratar el melanoma en pacientes con un tipo normal o salvaje. BRAF gene.
En 2015, la Administración de Drogas y Alimentos de los EE. UU. (FDA) aprobó cobimetinib (Cotellic ™, Genentech Inc. California, EE. UU.) En combinación con vemurafenib (una BRAF-quinasa específica inhibidor) para el tratamiento del melanoma.
Ese mismo año, el Comité de Medicamentos de Uso Humano de la Agencia Europea de Medicamentos emitió un dictamen positivo para la autorización de comercialización de cobimetinib en la Unión Europea.
Desde entonces, el cobimetinib recibió la aprobación para su comercialización en Nueva Zelanda a través de Medsafe en 2017 para el tratamiento de pacientes con melanoma.
La aprobación de la FDA se basó en los resultados del estudio de fase III CoBRIM, que mostró que cobimetinib, cuando se usa en combinación con vemurafenib, reduce el riesgo de empeoramiento de la enfermedad o muerte en aproximadamente la mitad en pacientes con melanoma.
Evidencia clave de ensayos clínicos para cobimetinib
El estudio CoBRIM
CoBRIM fue un internacional, aleatorizado, doble ciego, placebo-Estudio Fase III controlado que evaluó la seguridad y eficacia de 60 mg de cobimetinib una vez al día más 960 mg dos veces al día de vemurafenib en comparación con 960 mg de vemurafenib dos veces al día más placebo. Un total de 495 pacientes participaron en el estudio, todos los cuales no habían sido tratados previamente. BRAF V600 mutación-positivo, irresecable, localmente avanzado o metastásico melanoma.
La presencia de BRAF La mutación V600 se detectó utilizando el cobas®; 407 4800 BRAF Prueba de mutación V600.
- Todos los pacientes recibieron 960 mg de vemurafenib por vía oral dos veces al día en los días 1 a 28.
- Los pacientes fueron aleatorizados para recibir cobimetinib 60 mg o un placebo equivalente por vía oral una vez al día en los días 1 a 21 de cada ciclo de 28 días. Aleatorización se estratificó por región geográfica (América del Norte frente a Europa frente a Australia / Nueva Zelanda / otros) y estadio de la enfermedad (estadio IIIc irresecable, M1a o M1b frente al estadio M1c).
- El tratamiento se continuó hasta la progresión de la enfermedad, inaceptable toxicidad, o retiro del consentimiento.
- A los pacientes aleatorizados para recibir placebo no se les ofreció cobimetinib en el momento de la progresión de la enfermedad.
- El resultado de eficacia principal fue la supervivencia libre de progresión (SLP) evaluada por el investigador según RECIST v1.1.
- Los criterios de valoración secundarios incluyeron la SSP por un comité de revisión independiente, la tasa de respuesta objetiva, la supervivencia general y la duración de la respuesta.
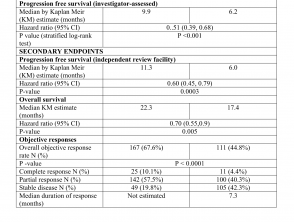
Los resultados de eficacia se resumen en la Tabla 1.
Resultados de eficacia del estudio CoBRIM
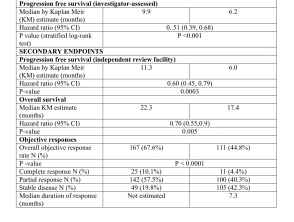
Resultados de eficacia del estudio CoBRIM
Eventos adversos – experiencia en ensayos clínicos (estudio CoBRIM)
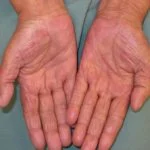
El más común (≥ 20%) Reacciones adversas con cobimetinib fueron:
- Diarrea
- Fotosensibilidad reacción
- Náusea
- Pirexia
- Vómitos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy diversas, reacción adversa Las tasas observadas en los ensayos clínicos pueden no reflejar las tasas observadas en la práctica.. En el estudio CoBRIM, el 15% de los pacientes que recibieron cobimetinib experimentaron una reacción adversa que resultó en la suspensión permanente del fármaco.
Las reacciones adversas más comunes que dieron lugar a la suspensión permanente del fármaco fueron:
- Laboratorio de hígado anormalidades, definido como aumentado aspartato aminotransferasa (2,4%), aumento de gamma glutamiltransferasa (1,6%) y aumento alanina aminotransferasa (ALT; 1,6%)
- Erupción (1,6%)
- Pirexia (1,2%)
- Desprendimiento de retina (2%).
Entre los 247 pacientes que recibieron cobimetinib, las reacciones adversas provocaron interrupciones o reducciones de la dosis en un 55%. Las razones más comunes para las interrupciones o reducciones de la dosis de cobimetinib fueron:
- Erupción (11%)
- Diarrea (9%)
- Coriorretinopatía – acumulación de líquido debajo de la retina (7%)
- Pirexia (6%)
- Vómitos (6%)
- Náuseas (5%)
- Aumento de la creatinfosfoquinasa (4,9%).
Incidencia de eventos adversos en> 10% de los pacientes (estudio CoBRIM)
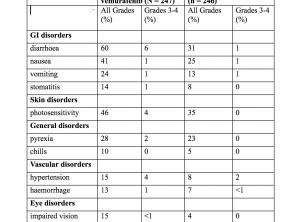
Eventos adversos (estudio CoBRIM)
Anormalidades en las pruebas de laboratorio (estudio CoBRIM)
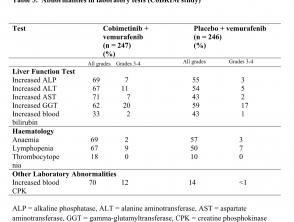
Anormalidades en las pruebas de laboratorio (estudio CoBRIM)
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas después de la autorización de un medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se recomienda a los profesionales sanitarios de Nueva Zelanda que notifiquen cualquier sospecha de reacciones adversas a el Centro de Farmacovigilancia de Nueva Zelanda.
Sobredosis
No hay experiencia con la sobredosis de cobimetinib en ensayos clínicos en humanos. En caso de sospecha de sobredosis, se debe suspender cobimetinib e instituir cuidados de apoyo. No existe un antídoto específico para la sobredosis de cobimetinib.
Se debe contactar al Centro Nacional de Venenos de Nueva Zelanda al 0800 764 766 para obtener asesoramiento sobre el manejo de la sobredosis.
Cobimetinib: potencial futuro
- Los nuevos tratamientos para el melanoma metastásico funcionan a través de distintos mecanismos para mejorar la respuesta inmunitaria y bloquear celular proliferación.
- Como trametinib, cobimetinib es un MEK inhibidor. MEK1 y MEK2 son tirosina quinasas que interactúan con BRAF y conducen a un crecimiento descontrolado de células de melanoma.
- Los ensayos clínicos han demostrado que la adición de cobimetinib a vemurafenib mejora la supervivencia en pacientes con melanoma que albergan la mutación V600 BRAF.
- La supervivencia global en pacientes tratados con nivolumab y pembrolizumab parece ser superior en comparación con lo logrado con cobimetinib y vemurafenib.
- Sin embargo, la supervivencia global en pacientes tratados con una combinación de vemurafenib y cobimetinib parece ser similar a otras estrategias de tratamiento disponibles para el tratamiento de metástasis irresecables. maligno melanoma como combinaciones de nivolumab e ipilimumab y dabrafenib con trametinib.
- Para la SLP, dabrafenib en combinación con trametinib y vemurafenib en combinación con cobimetinib parecían tener una mayor probabilidad de buen rendimiento que otras estrategias de tratamiento disponibles.
- Para los eventos adversos graves, pembrolizumab y nivolumab parecían tener una mayor probabilidad de menos eventos adversos graves que cobimetinib y vemurafenib.
- Sin embargo, ninguna de estas intervenciones es rentable a los precios minoristas máximos de farmacia, y los impactos presupuestarios en la práctica clínica pueden ser sustanciales.
- El rápido tumor respuesta a BRAF / MEK inhibición a menudo es de corta duración como tumores desarrollar resistencia a la terapia combinada.
- Nuevos ensayos están comenzando a investigar la adición de un tercer agente dirigido o inmunoterapia para aumentar la durabilidad de la respuesta al tratamiento.